
Amylose sous-type aa - causes, symptômes et traitement. Tout sur l'amylose - symptômes, diagnostic et traitement Avec les maladies purulentes chroniques prolongées, l'amyloïde se développe
RCHD (Centre républicain pour le développement de la santé du ministère de la Santé de la République du Kazakhstan)
Version : Protocoles cliniques du Ministère de la santé de la République du Kazakhstan - 2016
Amylose (E85)
Néphrologie
informations générales
Brève description
Approuvé
Commission mixte pour la qualité services médicaux
Ministère de la Santé et développement social République du Kazakhstan
du 13 octobre 2016
Protocole #13
Amylose- groupe de maladies poinçonner qui est le dépôt dans les tissus et les organes de glycoprotéine fibrillaire - amyloïde.
Corrélation entre les codes ICD-10 et ICD-9
| CIM-10 | CIM-9 | ||
| Code | Nom | Code | Nom |
| E85 | Amylose |
55.23 99.76 |
Biopsie rénale fermée [percutanée] [ponction]. Hémodialyse. Plasmaphérèse thérapeutique Immunoadsorption extracorporelle |
| E85.0 | Amylose familiale héréditaire sans neuropathie | ||
| E85.1 | Amylose héréditaire neuropathique | ||
| E85.2 | Amylose héréditaire, sans précision | ||
| E85.3 | Secondaire amylose systémique | ||
| E85.4 | Amylose limitée | ||
| E85.8 | Autres formes d'amylose | ||
| E85.9 | Amylose, sans précision | ||
Date de développement/révision du protocole : 2016.
Utilisateurs du protocole : médecins généralistes, thérapeutes, hématologues, néphrologues.
Échelle de niveau de preuve :
| UN | Méta-analyse de haute qualité, revue systématique d'ECR ou grands ECR avec une très faible probabilité (++) de biais dont les résultats peuvent être généralisés à une population appropriée. |
| DANS | Examen systématique de haute qualité (++) d'études de cohorte ou cas-témoins ou Etudes de cohorte ou cas-témoins de haute qualité (++) à très faible risque de biais ou ECR à faible (+) risque de biais, les résultats de qui peut être généralisée à la population appropriée. |
| AVEC |
Cohorte ou essai cas-témoins ou essai contrôlé sans randomisation à faible risque de biais (+). Dont les résultats peuvent être généralisés à la population appropriée ou aux ECR avec un risque de biais très faible ou faible (++ ou +), dont les résultats ne peuvent pas être directement généralisés à la population appropriée. |
| D | Description d'une série de cas ou d'une étude non contrôlée ou d'un avis d'expert. |
Classification
Types d'amyloïde et formes d'amylose correspondantes:
|
Protéine amyloïde |
Protéine précurseur | Formulaire clinique amylose |
| AA | Protéine SAA | Amylose secondaire dans les maladies chroniques maladies inflammatoires, y compris les maladies périodiques et le syndrome de Muckle-Wells |
| AL | Chaînes légères λ, κ des immunoglobulines | Amylose dans les dyscrasies plasmocytaires - idiopathiques, dans le myélome multiple et la macroglobulinémie de Waldenström |
| ATTR | Transthyrétine | Formes familiales d'amyloses polyneuropathiques, cardiopathiques et autres, amyloses séniles systémiques |
| Aβ2M | β2-microglobuline | amylose de dialyse |
| AGel | Gelsoline | Polyneuropathie amyloïde familiale finlandaise |
| AApoAI | Apolipoprotéine A-I | Polyneuropathie amyloïde(Type III, d'après van Allen, 1956) |
| fibrillation auriculaire | fibrinogène | néphropathie amyloïde |
| Aß | protéine β | Maladie d'Alzheimer, syndrome de Down, hémorragies cérébrales héréditaires avec amylose, Hollande |
| APrP Scr | Protéine prions | Maladie de Creutzfeldt-Jakob, maladie de Gerstmann-Straussler-Scheinker |
| AANF | Facteur natriurétique auriculaire | Amylose auriculaire isolée |
| AIAPP | Amyline | Amylose isolée des îlots de Langerhans diabète Type II, insuline |
| ACal | Procalcitonine | Pour le cancer médullaire glande thyroïde |
| ACys | Cystatine C | Hémorragies cérébrales héréditaires avec amylose, Islande |
Classification clinique de l'amylose
amylose primaire :
survenant sans raison apparente ;
Associé au myélome multiple
amylose secondaire:
dans les infections chroniques ;
dans la polyarthrite rhumatoïde et d'autres maladies tissu conjonctif;
dans les maladies oncologiques ;
amylose familiale (héréditaire):
en cas de maladie périodique;
variante portugaise et autres formes d'amylose familiale ;
amylose sénile
amylose locale
amylose héréditaire :
neuropathique
avec défaite membres inférieurs: portugais, japonais, suédois et autres types ;
avec défaite membres supérieurs: types Suisse-Indiana, Allemagne-Maryland;
néphropathique :
Maladie périodique
fièvre et douleurs abdominales chez les Suédois et les Siciliens ;
une combinaison d'éruptions cutanées, de surdité et de lésions rénales ;
Lésions rénales associées à une hypertension artérielle ;
cardiomyopathique :
Danois - insuffisance cardiaque progressive ;
· Mexicain-Américain - syndrome du sinus malade, arrêt auriculaire ;
mixte:
Finnois - dystrophie cornéenne et lésions des nerfs crâniens;
coups de cerveau.
Stades cliniques de l'amylose des reins
| Organiser | Manifestation clinique |
| 1 | Stade préclinique ou latent (asymptomatique) - l'amyloïde est présente dans la zone intermédiaire et un œdème et des foyers de sclérose se développent le long des vaisseaux directs des pyramides. L'étape dure 3 à 5 ans ou plus. Durant cette période, l'amylose réactive est dominée par manifestations cliniques maladie sous-jacente (par exemple, processus purulent dans les poumons, tuberculose, polyarthrite rhumatoïde etc.). |
| 2 | Stade protéinurique (albuminurique) - l'amyloïde apparaît principalement dans le mésangium, dans les boucles capillaires, dans les pyramides et cortex glomérule, dans les vaisseaux. La sclérose et l'atrophie des néphrons, l'hyperémie et la lymphostase se développent. Les reins sont élargis et denses, de couleur gris-rose terne. La protéinurie au début est modérément exprimée, elle peut même être transitoire pendant une certaine période, diminuer et augmenter, mais devient ensuite persistante (stade de protéinurie intermittente). Certains chercheurs distinguent deux périodes à ce stade : la protéinurie sélective et non sélective. La durée de l'étape est de 10 à 13 ans. |
| 3 | Stade néphrotique (œdémateux, œdémateux-hypotonique) - néphrose amyloïde-lipoïde - amyloïde dans toutes les parties du néphron. Il existe une sclérose et une amylose de la moelle, mais couche corticale sans exprimé changements sclérotiques. La durée de l'étape peut aller jusqu'à 6 ans. Tant au stade protéinurique qu'au stade néphrotique, les reins sont hypertrophiés, denses (gros rein sébacé). Cliniquement, ce stade se manifeste par le syndrome néphrotique classique avec tous ses signes : avec le développement protéinurie massive(avec une perte de protéines dans l'urine de plus de 3 à 5 grammes par jour), hypoprotéinémie avec hypoalbuminémie, hypercholestérolémie, lipidurie avec œdème jusqu'au degré d'anasarque. Des cylindres hyalins se trouvent dans le sédiment urinaire et, à mesure que la protéinurie augmente, des cylindres granulaires sont trouvés. Possible micro- et macrohématurie, leucocyturie sans signes de pyélonéphrite. |
| 4 | Stade urémique (terminal, azotémique) - rein ridé amyloïde - taille réduite, rein dense et cicatrisé. L'insuffisance rénale chronique diffère peu de celle des autres maladies rénales. On pense que contrairement à la glomérulonéphrite, dans laquelle l'apparition d'une IRC survenant avec une polyurie peut entraîner une convergence au moins partielle de l'œdème, dans l'amylose, l'azotémie se développe dans le contexte d'une pression artérielle basse et d'un syndrome néphrotique. |
Diagnostic (clinique externe)
DIAGNOSTIC AU NIVEAU AMBULATOIRE
Critères diagnostiques
Plaintes :
· faiblesse, fatigue;
· mal de tête;
gonflement des jambes, des bras et du visage ;
augmenté la pression artérielle;
nausées, diarrhée (diarrhée);
douleur dans la région du cœur;
douleur musculaire.
Anamnèse:
· perte de poids;
La présence d'une gammapathie monoclonale d'origine inconnue ;
maladies inflammatoires chroniques (purulentes);
infections chroniques;
hérédité.
Examen physique
Inspection générale:
Purpura périorbitaire (observé dans 15% des cas) ;
la macroglossie est caractéristique de l'amylose primaire (AL) ;
essoufflement quand activité physique(il y a environ 40% de patients);
signe de l'épaule (l'infiltration périarticulaire d'amyloïde entraîne une fausse hypertrophie et une augmentation du volume des muscles de la ceinture scapulaire et de la cuisse).
Auscultation:
présence possible d'arythmies cardiaques.
Palpation:
gonflement des membres inférieurs, dû à l'hypoalbuminémie et au syndrome néphrotique, ainsi qu'à la stagnation de grand cercle circulation due à une cardiomyopathie restrictive (observée dans 50% des cas) ;
hypertrophie du foie et de la rate;
paresthésie (observée chez environ 15 % des patients) ;
Douleur spastique dans le tractus gastro-intestinal ;
Il peut y avoir une augmentation des glandes salivaires sous-maxillaires.
Recherche en laboratoire :
Numération sanguine complète - anémie, leucocytose, augmentation de la VS ;
analyse générale d'urine - protéinurie, microhématurie, leucocyturie aseptique;
Test sanguin biochimique (protéines totales, albumine, Na, Ca, cholestérol, sucre dans le sérum sanguin) - hypoprotéinémie (due à l'hypoalbuminémie), hyperglobulinémie, hyponatrémie, hypoprothrombinémie, hypocalcémie, hypercholestérolémie.
Echographie des organes cavité abdominale et les reins - les reins compactés agrandis (gros reins gras) sont visualisés.

Diagnostic (hôpital)
DIAGNOSTIC AU NIVEAU STATIONNAIRE
Critères diagnostiques au niveau hospitalier
Plaintes et anamnèse: voir niveau ambulatoire.
Examen physique : voir niveau ambulatoire.
Recherche en laboratoire :
| test diagnostique | Résultat |
|
Immunofixation sérique Le test est positif chez 60 % des patients atteints d'amylose avec des immunoglobulines à chaîne légère (AL) (6). |
|
|
Immunofixation des urines Le test est positif chez 80 % des patients atteints d'amylose AL (6). La détection d'une protéine à chaîne légère dans l'urine suggère la présence d'un myélome multiple et d'une amylose. |
La présence d'une protéine monoclonale |
|
Test d'immunoglobuline à chaîne légère libre sérique Ce test relativement nouveau a une sensibilité très élevée (> 95 %) pour le diagnostic de l'amylose AL (10). Les antisérums disponibles dans le commerce contre les chaînes légères d'immunoglobuline, l'AA et la transthyrétine sont couramment utilisés mais peuvent ne pas avoir une spécificité et une sensibilité suffisantes. Dans de nombreux cas, la spectroscopie de masse et la microscopie immuno-électronique sont nécessaires pour déterminer le type d'amyloïde sous-jacent. |
Rapport kappa lambda anormal |
|
Biopsie moelle
La biopsie de la moelle osseuse est réalisée chez tous les patients suspectés d'amylose à chaînes légères et constitue une excellente source de tissu pour diagnostiquer tout patient suspecté d'amylose. |
Présence de plasmocytes clonaux |
Recherche instrumentale:
Échographie des organes abdominaux et des reins - des reins compactés élargis (gros reins gras) sont visualisés.
Algorithme diagnostique de l'amylose rénale
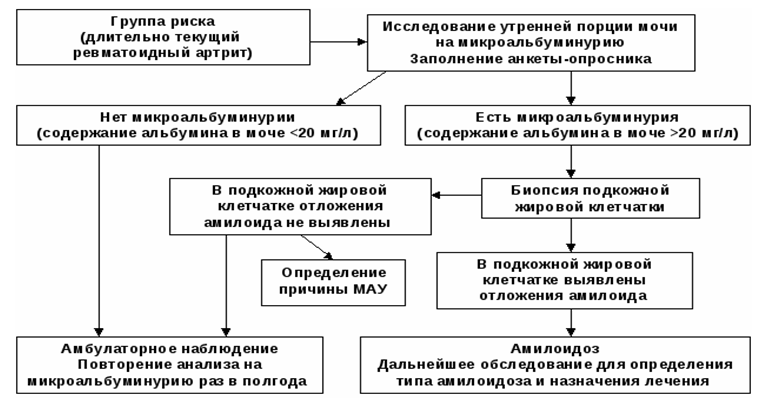
Liste des principaux mesures de diagnostic:
· analyse sanguine générale;
· analyse générale d'urine;
analyse biochimique du sang (protéines totales, albumine, Na, Ca, cholestérol, sucre dans le sérum sanguin);
immunofixation sérique;
immunofixation des urines ;
Enquête sur les immunoglobulines à chaîne légère libre dans le sérum ;
biopsie de la moelle osseuse.
Échographie des organes abdominaux et des reins.
Liste des mesures diagnostiques supplémentaires
Recherche en laboratoire :
| test diagnostique | Résultat |
|
Biopsie tissulaire : Pour le diagnostic de l'amylose, il est nécessaire que les dépôts dans les tissus du matériel de biopsie se colorent positivement pour le rouge Congo (11). Une biréfringence vert vif peut être observée lorsque le matériau Congo est coloré en rouge sous une lumière polarisée. Le matériel de biopsie peut être obtenu à partir de la muqueuse des lèvres, de la peau, des gencives, de la graisse sous-cutanée, de la moelle osseuse, des nerfs, du rectum, des reins, du foie ou du cœur. Les dépôts sont toujours situés en extracellulaire et sont amorphes. |
positif - biréfringence verte lorsqu'il est coloré avec du rouge Congo |
|
Etudes immunohistologiques des dépôts amyloïdes : Ils permettent de reconnaître Formes variées amylose systémique. |
antisérum contre l'immunoglobuline à chaîne légère, l'AA et la transthyrétine |
|
Masse - spectroscopie : Fournit une analyse de la composition des protéines amyloïdes. C'est actuellement l'étalon-or pour diagnostiquer le type d'amylose. |
confirme le type de protéine |
|
Microscopie immuno-électronique : Toutes les formes d'amyloïde au microscope électronique sont fibreuses, rigides et non ramifiées. |
les amyloïdes ont un aspect fibrillaire et sont rigides et non ramifiés. |
|
Test génétique: Les tests génétiques sont obligatoires pour exclure l'amylose héréditaire en présence de résultats douteux d'un test de détection d'immunoglobuline monoclonale/protéine de la chaîne légère libre de l'amyloïde. Les gènes peuvent être étudiés par séquençage direct et comprennent les gènes suivants : TTR, fibrinogène, apolipoprotéine A1, lysozyme, MEFV (fièvre méditerranéenne) et récepteur 1 du facteur de nécrose tumorale (TNFR1 ou TNFRSF1A). Le MEFV et le TNFRSF1A sont des syndromes de fièvre périodique héréditaire (c'est-à-dire des causes potentielles d'amylose AA) et ne sont pas des amyloses héréditaires en soi. |
positif |
|
Scintigraphie amyloïde P (SAP) sérique : DANS dernières années Dans la pratique clinique, la scintigraphie au composant P sérique marqué à l'iode (SAP) a commencé à être utilisée pour évaluer la distribution de l'amyloïde dans le corps. |
absorption aux sites de dépôt amyloïde |
|
Analyse générale sang: L'anémie s'observe principalement chez les patients insuffisance rénale ou saignement du tractus gastro-intestinal. La thrombocytémie est associée à une atteinte hépatique et à un hypersplénisme. |
généralement normal |
|
Analyse biochimique sang (tests hépatiques et rénaux), indicateurs d'état métabolique) : L'amylose hépatique est caractérisée par des taux élevés de phosphatase alcaline. Chez la majorité des patients sous stade précoce l'amylose des reins a préservé la clairance de la créatinine, mais il peut y avoir des degrés importants d'hypoalbuminémie en raison de la perte de protéines dans les urines (syndrome néphrotique). |
Faible taux d'albumine ; augmentation de la phosphatase alcaline |
|
Protéinurie quotidienne (collecte des urines en 24 heures) : L'excrétion d'albumine > 1 g/jour chez les patients atteints d'amylose indique une atteinte rénale (amylose rénale). Au niveau de protéinurie > 3 g/jour, un syndrome néphrotique se développe. |
protéine élevée dans les urines |
|
Niveau de troponine sérique : Un test sensible pour déterminer les dommages au myocarde. Les patients avec des niveaux de troponine détectables ont un pronostic plus mauvais que ceux qui n'en ont pas (12). |
élevé |
|
Peptide natriurétique de type B : sensible étude diagnostique pour distension myocardique et CHF. Il s'est avéré important valeur prédictive dans l'établissement de l'amylose du cœur (13). Au niveau du peptide natriurétique de type B > 300 ng/L (> 300 pg/mL) suggère une atteinte amyloïde myocardique (10). Les patients avec<170 нг / л (<170 пг / мл) имеют значительно более длительную выживаемость, чем пациенты с >170 ng/l (> 170 pg/ml). |
élevé |
|
bêta-2 microglobulines : C'est un facteur prédictif de survie chez les patients atteints d'amylose. Au niveau de bêta-2-microglobuline > 2,7 mg/l, le pronostic est défavorable (14). |
élevé |
Recherche instrumentale :
|
ECG : Doit être réalisée chez tous les patients dans le cadre d'une évaluation de l'atteinte cardiaque. |
trouble de la conduction du coeur |
|
Échocardiogramme (EchoCG): Signes cliniques l#39;insuffisance cardiaque chez les patients atteints d#39;amylose du cœur est observée de 22% à 34% (7). L'échocardiographie révèle une incidence élevée de dépôts amyloïdes chez les patients présentant des symptômes minimes (environ 50 % des cas d'AL ont une atteinte cardiaque). DANS dernière étape il y a une diminution de la fraction d'éjection. |
dysfonctionnement diastolique, épaississement septum interventriculaire, réduction de la fraction d'éjection |
|
Echo Doppler avec tension : Un indicateur du degré d'infiltration amyloïde dans le myocarde. A une grande sensibilité dans la détection des anomalies lorsqu'il n'y a pas hypertension artérielle ou une cardiopathie valvulaire. L'étirement myocardique est défini comme la variation en pourcentage de la longueur des fibres myocardiques par unité de longueur, et le taux dépend de la durée de l'étirement (15–16). |
Réduction de la contraction longitudinale et de l'étirement du myocarde ; restriction du remplissage ventriculaire |
|
IRM du coeur : La relaxométrie par résonance magnétique améliore la fiabilité du diagnostic IRM et aide à distinguer l'amylose cardiaque de la cardiomyopathie hypertrophique. |
augmentation significative des temps de relaxation T1 et T2 par rapport aux témoins d'âge |
Diagnostic différentiel
Diagnostic différentiel amylose rénale
| État | Signes/symptômes différenciables | Tests différentiables |
| Il est cliniquement difficile de distinguer la HCM de l'amylose cardiaque. |
l'échocardiographie est critère diagnostique pour le GCM, où une hypertrophie asymétrique du septum interventriculaire est détectée ; L'écho Doppler avec tension, utilisé pour exclure les signes d'amylose, n'indique pas les changements de remplissage restrictifs typiques détectés dans l'amylose; L'IRM permet de distinguer 2 syndromes |
|
| Glomérulopathie membraneuse | Cliniquement mêmes manifestations chez les patients atteints de syndrome néphrotique. | La biopsie rénale ne se colore pas avec le rouge Congo. |
| Neuropathie associée à la gammapathie monoclonale d'origine inconnue (MGNG) | Les patients ne présentent pas de degré significatif de protéinurie, d'hépatomégalie ou de cardiomyopathie. | la biopsie du nerf sural ne se colore pas au rouge Congo. |
| myélome multiple | Douleurs osseuses, symptômes d'anémie et d'insuffisance rénale. |
ordinaire radiographies présentent des lésions osseuses lytiques, des fractures par compression, une ostéoporose diffuse ; faible taux d'Hb ; insuffisance rénale. |
| le syndrome néphrotique | Protéinurie quotidienne supérieure à 3,5 g/jour, œdème, hypoalbuminémie, dyslipidémie | voir KP "Syndrome néphrotique" |
Traitement à l'étranger
Faites-vous soigner en Corée, en Israël, en Allemagne et aux États-Unis
Traitement à l'étranger
Obtenez des conseils sur le tourisme médical
Traitement
Drogues ( ingrédients actifs) utilisé dans le traitement
| Albumine humaine (Albumine humaine) |
| Anakinra (Anakinra) |
| Atorvastatine (atorvastatine) |
| Bortézomib (Bortézomib) |
| Valsartan (Valsartan) |
| Héparine sodique (héparine sodique) |
| Hydrochlorothiazide (Hydrochlorothiazide) |
| Dexaméthasone (Dexaméthasone) |
| Diflunisal (Diflunisal) |
| Indapamide (Indapamide) |
| Infliximab (Infliximab) |
| Canakinumab (Canakinumab) |
| Candésartan (candésartan) |
| Colchicine (Colchicine) |
| Lénalidomide (Lénalidomide) |
| Lisinopril (Lisinopril) |
| Losartan (Losartan) |
| Melphalan (Melphalan) |
| Nadroparine calcique (Nadroparine calcique) |
| Périndopril (Périndopril) |
| Ramipril (Ramipril) |
| Rilonacept (Rilonacept) |
| Rosuvastatine (rosuvastatine) |
| Simvastatine (Simvastatine) |
| Spironolactone (Spironolactone) |
| Thalidomide (Thalidomide) |
| Torasémide (Torasémide) |
| Fosinopril (Fosinopril) |
| Furosémide (furosémide) |
| Cyclophosphamide (Cyclophosphamide) |
| Étanercept (Étanercept) |
Traitement (ambulatoire)
TRAITEMENT AU NIVEAU AMBULATOIRE
Tactiques de traitement : si un diagnostic d'amylose rénale est suspecté, le patient doit être référé à un néphrologue pour un traitement ultérieur au niveau des patients hospitalisés.
Traitement non médicamenteux : Non.
Traitement médical: Non.
Autres types de traitement : Non.
Consultation de néphrologue - pour le diagnostic ;
consultation de spécialistes spécialisés en présence de pathologie concomitante.
Actions préventives:
Prévention primaire
Amylose rénale primaire mesures préventives Non;
Le développement d'une amylose secondaire à partir d'un état inflammatoire chronique est directement lié à une inflammation incontrôlée et à la synthèse de protéine amyloïde sérique par le foie. Le traitement de l'affection sous-jacente par la suppression de l'inflammation réduit le risque ultérieur d'amylose secondaire ;
Les patients atteints de gammapathie monoclonale inexpliquée connue sont à risque de développer une amylose, et une surveillance des patients est recommandée pour prévenir le développement d'une protéinurie, d'une neuropathie, d'une hépatomégalie ou d'une insuffisance cardiaque.
Traitement (ambulance)
DIAGNOSTIC ET TRAITEMENT AU STADE D'URGENCE
Mesures diagnostiques :
évaluation de l'état par un examen physique (mesure de la pression artérielle, de la fréquence cardiaque, auscultation).
Traitement médical: en présence de comorbidités, cf. protocole clinique selon les nosologies respectives.
Traitement (hôpital)
TRAITEMENT AU NIVEAU STATIONNAIRE
Tactiques de traitement :
Le traitement de l'amylose consiste à réduire la formation de protéines pathologiques et à protéger les organes de ses effets. Dans l'amylose AA, des mesures anti-inflammatoires sont utilisées en utilisant méthodes chirurgicales. À amylose secondaire traitement de la maladie sous-jacente. Dans l'amylose AL, la suppression des clones est réalisée plasmocytes qui synthétisent les immunoglobulines à chaîne légère. L'arrêt du dépôt d'immunoglobuline à chaîne légère permet au corps de dissoudre et d'éliminer l'excès d'amyloïde, ce qui empêche un dépôt ultérieur d'amyloïde. Pour les patients atteints d'amylose qui ont subi une biopsie et qui présentent un syndrome viscéral (c.-à-d. amyloïde dans le cœur, le foie, les reins, les nerfs, les poumons ou les intestins), une greffe de cellules souches/chimiothérapie est recommandée, qui est effectuée dans centre spécialisé pour le traitement de l'amylose.
Traitement non médicamenteux :
mode III: lit dans un état grave du patient et présence de complications, dosé activité physique, mode de vie sain vie, arrêter de fumer et boire de l'alcool;
· Régime : n° 7. Apport protéique équilibré et suffisant (1,5-2g/kg), calories selon l'âge, en présence d'œdèmes et d'hypertension - limitant l'utilisation de chlorure de sodium ( sel de table) < 1-2г/сут;
surveillance du niveau de protéinurie par bandelettes de test 1 fois en 1-2 semaines, mesure régulière de la pression artérielle.
En cas d'augmentation de la protéinurie (rechute), détermination du rapport protéines/créatinine (pour calculer la protéinurie quotidienne) et correction thérapie pathogénique;
En cas de résistance au traitement immunosuppresseur en cours, correction du traitement en milieu hospitalier.
Traitement médical: Le traitement médical de l'amylose des reins est l'utilisation de la chimiothérapie en conjonction avec la greffe de cellules souches.
Type d'amylose AL.
Diagnostiqué pour la première foisALamylose :
Chimiothérapie myéloablative à fortes doses de melphalan* et de TSC ;
Chimiothérapie myéloablative avec de fortes doses de melphalan* (après enregistrement en République du Kazakhstan) et TSC en thérapie d'induction avec le bortézomib associé à la dexaméthasone ;
Indications pour le TSC :
· âge<70 лет;
Contre-indications pour le TSC:
Insuffisance cardiaque grave
bilirubine totale > 51 µmol/l (> 3 mg/dl) ;
fraction d'éjection d'écho<45%;
Troponine sérique >0,1 µg/L (>0,1 ng/mL).
NB ! La condition standard pour la transplantation est une dose unique de melphalan*. Il est généralement administré en fonction du risque à des doses allant de 140 mg/m 2 pour les patients à risque intermédiaire à 200 mg/m 2 pour les patients à faible risque. La récolte de cellules souches implique l'utilisation de facteurs de croissance uniquement. La collecte minimale de cellules souches doit être de 3x10 6 cellules CD34 par kg de poids du patient.
NB ! Les patients peuvent également recevoir un traitement d'induction avec le bortézomib plus la dexaméthasone avant le TSC.
Avec une réponse incomplète au TSC
Chimiothérapie après TSC
Pour les patients qui n'atteignent pas des niveaux normalisés de chaînes légères libres d'immunoglobulines, une combinaison de melphalan* et de dexaméthasone/cyclophosphamide, dexaméthasone et thalidomide* est recommandée. Les cycles sont répétés mensuellement jusqu'à 1 an.
Indications pour ajuster la dose du régime CDT :
âge > 70 ans ;
insuffisance cardiaque supérieure à NYHA II ;
avec une surcharge importante de liquide corporel.
NB ! Les patients TSC partiellement répondeurs deviennent totalement répondeurs après un traitement adjuvant par la thalidomide* et la dexaméthasone. la thalidomide* est assez toxique chez les patients sensibles et aucune tolérance ne se développe chez les patients à la dose suggérée de 200 mg/jour, qui est couramment utilisée chez les patients atteints de myélome multiple. Les patients atteints d'amylose ne peuvent généralement pas tolérer des doses de thalidomide* > 50 mg/jour. La thalidomide* provoque des symptômes neurologiques, constipation, éruption cutanée et somnolence. La thérapie ne dépasse généralement pas un an.
Les principales associations médicamenteuses sont le melphalan* avec dexaméthasone/cyclophosphamide + dexaméthasone + thalidomide*.
Si la TSC est inefficace, une chimiothérapie est en outre recommandée :
melphalan* et dexaméthasone pendant 6 à 12 mois/bortézomib et dexaméthasone pour le bortézomib une semaine et dexaméthasone la semaine suivante jusqu'à 45 semaines/bortézomib en monothérapie.
En cas d'échec d'un traitement antérieur par bortézomib, un traitement par lénalidomide plus dexaméthasone peut être envisagé.
Les principales combinaisons de médicaments sont le melphalan* avec la Dexaméthasone/Bortézomib avec la Dexaméthasone/Bortézomib en monothérapie Combinaisons alternatives médicaments sont le lénalidomide avec la dexaméthasone.
Amylose AL non soumise à TSC.
Premier diagnostic.
Chimiothérapie:
L'association du melphalan* à la dexaméthasone est la principale option thérapeutique (UD-B) ;
Les autres médicaments comprennent le cyclophosphamide, la dexaméthasone et la thalidomide* (UD-C)/lénalidomide avec dexaméthasone (UD-B) ;
· La monothérapie à la dexaméthasone peut être administrée aux patients sensibles au traitement au melphalan* (UD-B).
Indications pour le TSC :
· âge<70 лет;
signes minimes d'insuffisance cardiaque (NYHA
Atteinte de moins de 3 organes par l'amyloïde.
NB ! A ce stade, le dosage de la chaîne légère libre des immunoglobulines est recommandé pour évaluer l'efficacité du traitement et déterminer la durée du traitement (de 6 à 12 mois).
NB ! Les principales associations médicamenteuses sont le melphalan* avec la Dexaméthasone.
Les associations médicamenteuses alternatives sont cyclophosphamide + dexaméthasone + thalidomide*/lénalidomide + dexaméthasone/dexaméthasone en monothérapie.
NB ! En cas d'inefficacité partielle du premier cycle de chimiothérapie, un traitement par Bortezomib est recommandé.
Si une rechute survient après le traitement principal :
Répéter les cures de chimiothérapie
des cures mensuelles de melphalan* et de dexaméthasone, de cyclophosphamide pendant 6 à 12 mois, dexaméthasone et thalidomide* (CDT), de lénalidomide avec dexaméthasone mensuellement pendant une durée indéterminée sont recommandées / des cures de bortézomib et de dexaméthasone doivent être envisagées.
Les principales associations médicamenteuses sont melphalan* avec dexaméthasone/cyclophosphamide + dexaméthasone + thalidomide*/lénalidomide avec dexaméthasone/dexaméthasone avec bortézomib.
Traitement de l'amylose de type AA.
Traitement de la maladie sous-jacente :
le traitement comprend un contrôle complet du processus inflammatoire systémique sous-jacent ;
· Les arthropathies inflammatoires sont traitées par infliximab et étanercept avec une durée médiane de traitement de 20 mois. Le blocage de l'interleukine-1 est possible avec l'inefficacité / le refus du patient de l'infliximab ou de l'étanercept ;
Médicaments essentiels infliximab/étanercept. De plus, l'anakinra*, le canakinumab ou le rilonacept* peuvent être utilisés.
NB ! Si l'amylose est due à une forme localisée de la maladie de Castleman, la résection de la tumeur est une méthode efficace.
Amylose secondaire familiale
Fièvre méditerranéenne familiale la colchicine* 0,5 à 0,6 mg deux fois par jour est recommandée.
Forme d'amylose à transthyrétine :
Le diflunisal* ralentit la progression de la neuropathie dans les formes mutantes multiples d'amylose à transthyrétine. Tafamidis retarde la progression de la neuropathie dans l'amylose héréditaire à transthyrétine Val30Met. Les principaux médicaments sont le diflunisal* ou le tafamidis*.
Médicaments chimiothérapeutiques
Des anticorps monoclonaux
· l'introduction d'anticorps appelés 11-1f4 induit une réponse inflammatoire à médiation cellulaire facilitée, entraînant une diminution rapide de l'amyloïde. L'étanercept est recommandé pour le traitement des patients atteints d'AL avancée (LE-C) ;
L'éprodisate* réduit le risque d'insuffisance rénale liée à la dialyse chez les patients atteints d'amylose AA en déstabilisant le squelette glycosaminoglycane des fibrilles amyloïdes.
Traitement de l'amylose de type AL
| Les préparatifs | une seule dose | Multiplicité d'introduction |
| Melphalan* | 140-200mg/m2 | une fois |
| Bortézomide | 1,3 mg/m2 | 2 fois par semaine selon le schéma |
| Dexaméthasone | 40 mg/jour | 1 fois par jour par voie orale ou intraveineuse selon le schéma |
| Cyclophosphamide | 10mg/kg | 1 jour i/v |
|
Thalidomide* |
200 mg/jour |
1 fois par jour de préférence au coucher et au moins 1 heure après les repas |
| Lénalidomide | 25 mg/jour | 1 fois par jour selon le schéma |
Traitement de l'amylose de type AA
| Les préparatifs | une seule dose | Multiplicité d'introduction |
| l'infliximab | 3-10 mg/jour | 1 fois par jour in/in selon le schéma |
| étanercept | 50mg | 1 fois par semaine s/c |
| Anakinra* | 100mg | 1 fois par jour s/c |
| Canakinumab | 150-300mg | 1 fois en 4 semaines s/c selon le schéma |
| Rilonacept* | 320 mg/jour | 160 mg s / c dans différentes zones |
Amylose secondaire familiale
Liste des médicaments principaux et complémentaires :
Liste des médicaments essentiels :
anakinra* ;
bortézomide;
dexaméthasone;
diflunisal*;
l'infliximab ;
colchicine* ;
lénalidomide;
melphalan*;
· rilonacept* ;
thalidomide* ;
canakinumab ;
· cyclophosphamide ;
étanercept.
Liste des médicaments supplémentaires :
| Thérapie néphroprotectrice - inhibiteurs de l'enzyme de conversion de l'angiotensine | ||
| Les préparatifs | une seule dose | Multiplicité d'introduction |
|
Lisinopril Ramipril Fosinopril Périndopril |
5 - 10 mg 5 - 10 mg 5 - 10 mg 2,5 à 5 mg |
1-2 fois 1-2 fois 1-2 fois 1-2 fois |
| Thérapie néphroprotectrice - antagonistes de la rénine-angiotensine II | ||
|
Losartan Valsartan Candésartan |
50-100 mg 80-160mg 8 - 16 mg |
1-2 fois 1-2 fois 1-2 fois |
| Diurétiques | ||
|
En boucle : furosémide torasémide De type thiazidique : hypothiazide indapamide Antagonistes de l'aldostérone spironolactone |
1-3mg/kg/jour 5-10 mg 25-100 mg 12.5-25mg/jour |
Une fois Une fois Une fois |
| Anticoagulants | ||
| Héparine sodique Nadroparine calcique Énoxaparine sodique |
2500-5000 UI 1000-5000 UI 1000-5000 UI |
1 à 2 fois par jour 1 à 2 fois par jour 1 à 2 fois par jour |
| Statines | ||
|
Rosuvastatine Simvastatine Atorvastatine |
10-20 mg 10-20 mg 10-20 mg |
1 par jour 1 par jour 1 par jour |
| Substituts du plasma et autres composants sanguins | ||
| Albumen | 10 % 200 ml, 20 % 100 ml | sur demande |
NB ! *l'utilisation du médicament après son enregistrement en République du Kazakhstan
Intervention chirurgicale:
Greffe de rein de donneur.
Les indications:
Le développement de l'insuffisance rénale chronique;
· HGN.
Séquestrectomie
les indications:
ostéomyélite,
Ablation d'un lobe pulmonaire
les indications:
maladie de la bronchectasie.
greffe de cellules souches
Indications pour le TSC :
Moins de 70 ans
avec une insuffisance cardiaque minime (classe
Implication de moins de 3 organes dans le processus.
NB ! La chimiothérapie myéloablative au melphalan* suivie d'une greffe de cellules souches pour la guérison est indiquée chez les patients à faible risque de complications liées au traitement (LE-A). Les avantages de la transplantation pour l'amylose n'ont pas été prouvés de manière fiable (LE-B). La TSC est la seule thérapie disponible dans ce cas.
Complications du SCT :
· mort cardiaque subite;
saignement du tractus gastro-intestinal;
insuffisance rénale.
Autres types de traitement :
Thérapie de remplacement rénal (hémodialyse, hémodiafiltration, dialyse péritonéale).
Indications pour un avis d'expert : consultation de spécialistes spécialisés en présence de pathologie concomitante.
Indications de transfert en réanimation et réanimation :
complication incontrôlée du syndrome néphrotique et de l'IRA ;
manifestation extrarénale d'amylose nécessitant une hospitalisation en réanimation.
Indicateurs d'efficacité du traitement
Stabilisation / restauration de la fonction des organes vitaux ;
la prévention des troubles fonctionnels, avec une augmentation de l'espérance de vie des patients ;
régression du syndrome néphrotique;
réduction de la protéinurie ;
Réduction des dépôts amyloïdes dans les tissus.
Gestion complémentaire:
surveillance ambulatoire d'un spécialiste au lieu de résidence;
Échographie des reins 1 fois en 3 mois ;
Tests sanguins, tests d'urine 1 fois pendant 3 mois.
Hospitalisation
Indications d'hospitalisation programmée :
vérification du diagnostic d'amylose des reins;
la présence d'un syndrome néphrotique.
Indications d'hospitalisation d'urgence:
Anasarque (gonflement diffus des tissus mous avec localisation prédominante dans la moitié inférieure du corps) ;
oligoanurie (une forte diminution de la quantité d'urine excrétée par les reins).
Information
Sources et littérature
- Procès-verbaux des réunions de la Commission mixte sur la qualité des services médicaux du MHSD RK, 2016
- Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Définition de l'atteinte d'organes et de la réponse au traitement dans l'amylose à chaîne légère d'immunoglobuline (AL) : avis consensuel du 10e Symposium international sur l'amyloïde et l'amylose, Tours, France, 18-22 avril 2004. Am J Hematol. 2005;79(4):319-28. 2) Critères de classification des gammapathies monoclonales, du myélome multiple et des troubles apparentés : un rapport du Groupe de travail international sur le myélome. Br J Haematol. 2003;121(5):749-57. 3) BMJ Best Practice : Amylose : BMJ Publishing Group ; 2016 . Disponible sur : http://bestpractice.bmj.com/best-practice/monograph/444.html. 4) Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, et al. Amyloïde : vers une clarification terminologique. Rapport du comité de nomenclature de la Société internationale de l'amylose. amyloïde. 2005;12(1):1-4. 5) Lignes directrices cliniques nationales pour le diagnostic et le traitement de l'amylose AA et AL. Société scientifique des néphrologues de Russie. 2016 . Disponible sur : http://nonr.ru/?page_id=3178. 6) Kyle RA, Gertz MA. Amylose systémique primitive : caractéristiques cliniques et biologiques de 474 cas. Sémin Hématol. 1995;32(1):45-59. 7) Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR. amylose. Best Pract Res Clin Haematol. 2005;18(4):709-27. 8) Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR. Amylose : diagnostic et prise en charge. Clin Lymphome Myélome. 2005;6(3):208-19. 9) Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. Une étude à long terme du pronostic dans la gammapathie monoclonale de signification indéterminée. N Engl J Méd. 2002;346(8):564-9. 10) Palladini G, Russo P, Bosoni T, Verga L, Sarais G, Lavatelli F, et al. L'identification des chaînes légères amyloïdogènes nécessite la combinaison d'un dosage des chaînes légères sans sérum avec une immunofixation du sérum et de l'urine. Clinique Chim. 2009;55(3):499-504. 11) Gertz MA La classification et le typage des dépôts amyloïdes. Suis J Clin Pathol. 2004;121(6):787-9. 12) Dispenzieri A, Kyle RA, Gertz MA, Therneau TM, Miller WL, Chandrasekaran K, et al. Survie chez les patients atteints d'amylose systémique primaire et de troponines cardiaques sériques élevées. Lancette. 2003;361(9371):1787-9. 13) Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, Perfetti V, et al. Le peptide natriurétique pro-cerveau N-terminal sérique est un marqueur sensible du dysfonctionnement myocardique dans l'amylose AL. circulation. 2003;107(19):2440-5. 14) Zerbini CA, Anderson JJ, Kane KA, Ju ST, Campistol JM, Simms RW, et al. Niveaux sériques de bêta 2 microglobuline et prédiction de la survie dans l'amylose AL. amyloïde. 2002;9(4):242-6. 15) Koyama J, Ray-Sequin PA, Falk RH. Fonction myocardique longitudinale évaluée par la vitesse tissulaire, la tension et l'échocardiographie Doppler tissulaire chez les patients atteints d'amylose cardiaque AL (primaire). circulation. 2003;107(19):2446-52. 16) Weidemann F, Strotmann JM. Utilisation de l'imagerie Doppler tissulaire pour identifier et gérer les maladies systémiques. Clin Res Cardiol. 2008;97(2):65-73. 17) Comenzo RL, Gertz MA. Transplantation autologue de cellules souches pour l'amylose systémique primaire. Sang. 2002;99(12):4276-82. 18) Huang X, Wang Q, Chen W, Zeng C, Chen Z, Gong D, et al. Thérapie d'induction avec bortézomib et dexaméthasone suivie d'une autogreffe de cellules souches par rapport à une autogreffe de cellules souches seule dans le traitement de l'amylose rénale AL : un essai contrôlé randomisé. BMC Med. 2014;12:2. 19) Cohen AD, Zhou P, Chou J, Teruya-Feldstein J, Reich L, Hassoun H, et al. Transplantation autologue de cellules souches adaptée au risque avec adjuvant dexaméthasone +/- thalidomide pour l'amylose systémique à chaîne légère : résultats d'un essai de phase II. Br J Haematol. 2007;139(2):224-33. 20) Mahmood S, Venner CP, Sachchithanantham S, Lane T, Rannigan L, Foard D, et al. Lénalidomide et dexaméthasone pour l'amylose AL systémique après un traitement antérieur avec des schémas thérapeutiques à base de thalidomide ou de bortézomib. Br J Haematol. 2014;166(6):842-8. 21) Gertz MA, Lacy MQ, Lust JA, Greipp PR, Witzig TE, Kyle RA. Essai de phase II de la dexaméthasone à haute dose pour les patients non traités atteints d'amylose systémique primaire. Med Oncol. 1999;16(2):104-9. 22) ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommandations pour la prise en charge des maladies auto-inflammatoires. Ann Rheum Dis. 2015;74(9):1636-44. 23) Lidar M, Livneh A. Fièvre méditerranéenne familiale : progrès cliniques, moléculaires et de gestion. Neth J Med. 2007;65(9):318-24. 24) Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Réutilisation du diflunisal pour la polyneuropathie amyloïde familiale : un essai clinique randomisé. JAMA. 2013;310(24):2658-67. 25) Coelho T, Maia LF, da Silva AM, Cruz MW, Plante-Bordeneuve V, Suhr OB, et al. Effets à long terme du tafamidis pour le traitement de la polyneuropathie amyloïde familiale à transthyrétine. J Neurol. 2013;260(11):2802-14. 26) Zhang KY, Tung BY, Kowdley KV. Transplantation hépatique pour les maladies hépatiques métaboliques. Clin Foie Dis. 2007;11(2):265-81. 27) Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar S. greffe pour amylose. Opinion actuelle sur le col. 2007;19(2):136-41. 28) Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. Melphalan à haute dose versus melphalan plus dexaméthasone pour l'amylose AL. N Engl J Méd. 2007;357(11):1083-93. 29) Mhaskar R, Kumar A, Behera M, Kharfan-Dabaja MA, Djulbegovic B. Rôle de la chimiothérapie à haute dose et de la greffe de cellules hématopoïétiques autologues dans l'amylose systémique primaire : une revue systématique. Greffe de Moelle Sanguine Biol. 2009;15(8):893-902. 30) Gertz MA Amylose à chaîne légère d'immunoglobuline : mise à jour 2014 sur le diagnostic, le pronostic et le traitement. Suis J Hematol. 2014;89(12):1132-40. 31) Sitia R, Palladini G, Merlini G. Bortezomib dans le traitement de l'amylose AL : thérapie ciblée ? hématologique. 2007;92(10):1302-7. 32) Kastritis E, Anagnostopoulos A, Roussou M, Toumanidis S, Pamboukas C, Migkou M, et al. Traitement de l'amylose à chaîne légère (AL) avec l'association de bortézomib et de dexaméthasone. hématologique. 2007;92(10):1351-8. 33) Palladini G, Russo P, Nuvolone M, Lavatelli F, Perfetti V, Obici L, et al. Le traitement par melphalan oral plus dexaméthasone produit des rémissions à long terme dans l'amylose AL. Sang. 2007;110(2):787-8. 34) Wechalekar AD, Goodman HJ, Lachmann HJ, Offer M, Hawkins PN, Gillmore JD. Innocuité et efficacité du cyclophosphamide, de la thalidomide et de la dexaméthasone adaptés au risque dans l'amylose AL systémique. Sang. 2007;109(2):457-64. 35) Dispenzieri A, Lacy MQ, Zeldenrust SR, Hayman SR, Kumar SK, Geyer SM, et al. L'activité du lénalidomide avec ou sans dexaméthasone chez les patients atteints d'amylose systémique primaire. Sang. 2007;109(2):465-70. 36) Moreau P, Jaccard A, Benboubker L, Royer B, Leleu X, Bridoux F, et al. Lénalidomide en association avec le melphalan et la dexaméthasone chez les patients atteints d'amylose AL nouvellement diagnostiquée : une étude multicentrique de phase 1/2 à dose croissante. Sang. 2010;116(23):4777-82. 37) Reece DE, Sanchorawala V, Hegenbart U, Merlini G, Palladini G, Fermand JP, et al. Bortézomib hebdomadaire et bihebdomadaire chez les patients atteints d'amylose AL systémique : résultats d'une étude de phase 1 à doses croissantes. Sang. 2009;114(8):1489-97. 38) Kastritis E, Wechalekar AD, Dimopoulos MA, Merlini G, Hawkins PN, Perfetti V, et al. Bortézomib avec ou sans dexaméthasone dans l'amylose systémique primaire (chaîne légère). J Clin Oncol. 2010;28(6):1031-7. 39) Lamm W, Willenbacher W, Lang A, Zojer N, Muldur E, Ludwig H, et al. Efficacité de l'association du bortézomib et de la dexaméthasone dans l'amylose AL systémique. Anne Hématol. 2011;90(2):201-6. 40) Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, et al. Eprodisate pour le traitement de la maladie rénale dans l'amylose AA. N Engl J Méd. 2007;356(23):2349-60. 41) Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Les anticorps dirigés contre le composant amyloïde P du sérum humain éliminent les dépôts amyloïdes viscéraux. Nature. 2010;468(7320):93-7. 42) Produits pharmaceutiques M. Étude sur la dexaméthasone plus IXAZOMIB (MLN9708) ou le choix du traitement par les médecins dans l'amylose systémique à chaînes légères (AL) récidivante ou réfractaire (NCT01659658) février 2016 . Disponible sur : https://clinicaltrials.gov/ct2/show/NCT01659658. 43) Salomon A, Weiss DT, Wall JS. Potentiel thérapeutique de l'anticorps monoclonal chimérique réactif à l'amyloïde 11-1F4. Clinique Cancer Res. 2003;9(10 Pt 2):3831S-8S. 44) Solomon A, Weiss DT, Wall JS. Immunothérapie dans l'amylose primaire systémique (AL) utilisant des anticorps monoclonaux réactifs à l'amyloïde. Cancer Biother Radiopharm. 2003;18(6):853-60. 45) Hussein MA, Juturi JV, Rybicki L, Lutton S, Murphy BR, Karam MA. Traitement à l'étanercept chez les patients atteints d'amylose primaire avancée. Med Oncol. 2003;20(3):283-90. 46) Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Innocuité et efficacité de la thérapie ARNi pour l'amylose à transthyrétine. N Engl J Méd. 2013;369(9):819-29. 47) Rajkumar SV, Lacy MQ, Kyle RA. Gammapathie monoclonale de signification indéterminée et myélome multiple indolent. Blood Rev. 2007;21(5):255-65.
Information
Abréviations utilisées dans le protocole
| FAV | fistule artério-veineuse |
| SA | hypertension artérielle |
| ENFER | la pression artérielle |
| bkk | bloqueurs de canaux calciques |
| BRA | bloqueurs des récepteurs de l'angiotensine |
| BRV | survie sans maladie |
| I/V | administration intraveineuse |
| MCG | Cardiomyopathie hypertrophique |
| DBP | maladie rénale diabétique |
| DN | néphropathie diabétique |
| tube digestif | tube digestif |
| RRT | thérapie de remplacement rénal |
| Inhibiteur de l'ECA | inhibiteurs de l'enzyme de conversion de l'angiotensine |
| PC | protocole clinique |
| MGNG | Gammapathie monoclonale d'origine inconnue |
| UI | unité internationale |
| CIM | Classification internationale des maladies |
| ARNm | matrice acide ribonucléique |
| IRM | - Imagerie par résonance magnétique |
| N.-É. | le syndrome néphrotique |
| OB | la survie globale |
| Police provinciale de l'Ontario | lésion rénale aiguë |
| PC | sous-cutané |
| DFG | Taux de filtration glomérulaire |
| CST | greffe de cellules souches |
| DU | un niveau de confiance |
| ultrason | échographie |
| MRC | maladie rénale chronique |
| FRC | l'insuffisance rénale chronique |
| CHF | Insuffisance cardiaque chronique |
| rythme cardiaque | taux d'insuffisance cardiaque |
| CDT | cyclophosphamide, dexaméthasone, thalidomide |
| échocardiographie | échocardiogramme |
| Hb | hémoglobine |
| NYHA | New York Heart Association MEFV - Fièvre méditerranéenne (fièvre méditerranéenne) |
| SÈVE | Composant amyloïde P sérique (composant P de l'amyloïde sérique) |
| TTR | transtéritine (transtéritine) |
| TNFR1 | récepteur 1 du facteur de nécrose tumorale |
| TNFRSF1A | superfamille des récepteurs du facteur de nécrose tumorale, membre 1A |
Liste des développeurs de protocoles avec des données de qualification :
1) Tuganbekova Saltanat Kenesovna - Docteur en sciences médicales, professeur au JSC "Centre médical scientifique national", thérapeute en chef, néphrologue indépendant en chef du ministère de la Santé et du Développement social de la République du Kazakhstan.
2) Gaipov Abdujapar Erkinovich - Candidat en sciences médicales, professeur agrégé, chef du département d'hémocorrection extracorporelle de JSC NSMC, néphrologue de première catégorie, secrétaire général de l'ONG "Society of Nephrologists, Dialysis Physicians and Transplantologists".
3) Turebekov Duman Kazhibaevich - Docteur en sciences médicales, professeur agrégé, néphrologue en chef du département de la santé d'Astana, néphrologue de première catégorie, chef du département de néphrologie et de thérapie de l'hôpital municipal n ° 1 d'Astana.
4) Khudaibergenova Mahira Seidualievna - expert en chef en pharmacologie clinique du JSC "Centre scientifique national d'oncologie et de transplantation".
Indication d'absence de conflit d'intérêts : Non.
Liste des examinateurs : Ainabekova Bayan Alkenovna - Docteur en sciences médicales, professeur, chef du département de médecine interne pour stage et résidence, JSC "Astana Medical University".
Fichiers joints
Attention!
- En vous soignant, vous pouvez causer des dommages irréparables à votre santé.
- Les informations publiées sur le site Web de MedElement ne peuvent et ne doivent pas remplacer une consultation médicale en personne. Assurez-vous de contacter les installations médicales si vous avez des maladies ou des symptômes qui vous dérangent.
- Le choix des médicaments et leur posologie doivent être discutés avec un spécialiste. Seul un médecin peut prescrire le bon médicament et sa posologie, en tenant compte de la maladie et de l'état du corps du patient.
- Le site Web MedElement est une ressource d'information et de référence uniquement. Les informations publiées sur ce site ne doivent pas être utilisées pour modifier arbitrairement les prescriptions du médecin.
- Les éditeurs de MedElement déclinent toute responsabilité en cas d'atteinte à la santé ou de préjudice matériel résultant de l'utilisation de ce site.
Le terme «amylose» est retenu par respect pour Rudolf Virchow, qui a été le pionnier de l'utilisation des techniques de coloration histochimique en 1854 pour caractériser les dépôts amyloïdes dans des échantillons cérébraux pathologiques. Alors que toutes les autres structures de ses sections de cerveau se sont colorées en jaune après l'ajout d'iode et d'acide sulfurique, les corps amyloïdes se sont colorés en bleu clair avec de l'iode et en violet brillant avec l'ajout ultérieur d'acide. Parce que ce type de coloration était caractéristique de la cellulose végétale, Virchow a conclu que les corps amyloïdes étaient composés d'une substance similaire à la cellulose, qu'il a appelée amyloïde. Le terme « amyloïde » signifie « contenant » ou « ressemblant à de l'amidon ». Cependant, il s'agit d'un terme incorrect, car on sait maintenant que les dépôts amyloïdes contiennent principalement des protéines, même si certaines substances contenant des glucides peuvent se lier aux protéines. La recherche sur l'amyloïde s'est principalement concentrée sur sa composition protéique.
Le début et la progression de l'amyloïdogenèse dépendent entièrement de la protéine responsable, mais suivent généralement l'un des trois processus pathogènes suivants : surproduction et dépôt d'une protéine de type sauvage, dépôt d'un variant de protéine mutée ou dépôt de fragments de protéine qui ont été générés par clivage endoprotéolytique aberrant.
La présence d'amyloïde détectable est une condition préalable à la manifestation de la maladie chez les patients. Bien que l'étendue et le taux des dommages aux organes et la gravité de la maladie varient d'un patient à l'autre, même chez ceux qui ont des types similaires de protéines amyloïdes, la charge amyloïde totale du corps est directement corrélée à la gravité de la maladie. Ainsi, la réduction de la quantité totale d'amyloïde peut stabiliser ou améliorer les manifestations cliniques de la maladie.
Prévalence
La prévalence de l'amylose varie selon les régions. Bien que la maladie d'Alzheimer soit la forme d'amylose la plus courante aux États-Unis et dans le monde, nous nous sommes principalement concentrés sur les formes systémiques de la maladie. Aux États-Unis, l'AL est la forme la plus courante d'amylose systémique. Parmi les résidents du comté d'Olmsted, Minnesota, des données fiables ont été obtenues concernant la prévalence de la maladie entre 1950 et 1989. Selon ces informations, environ 1 personne sur 100 000 développera une amylose AL.
Dans le monde, l'AA est la forme la plus courante d'amylose. Dans les pays industrialisés, les maladies inflammatoires sont la principale cause d'amylose AA, tandis que les infections systémiques ou chroniques sont responsables de la plupart des cas d'amylose AA dans les pays en développement.
L'amylose peut se présenter comme une maladie systémique ou localisée. Il existe quatre classes d'amylose systémique : AL, AA, ATTR et Ap2M. De nombreuses formes d'amylose localisée ont été identifiées. La maladie d'Alzheimer et les dépôts amyloïdes localisés dans le larynx et les voies urinaires sont les formes les plus courantes d'amylose localisée.
À l'exception de la maladie d'Alzheimer, dans laquelle il existe un effet cytotoxique sur les cellules cérébrales, le tableau clinique des autres amyloses, comme décrit précédemment, est causé par une perturbation mécanique de la fonction physiologique normale. Les manifestations cliniques de l'amylose dépendent du type de protéine amyloïde.
Amylose-AL
Les manifestations cliniques de l'amylose AL sont différentes. Les reins, le cœur et le foie sont les organes les plus fréquemment et les plus sensiblement touchés ; cependant, tous les organes autres que le système nerveux central peuvent être affectés. Dans les reins, les dépôts d'amyloïde AL sont principalement observés dans les glomérules, ce qui provoque un syndrome néphrotique, qui se manifeste généralement par une excrétion initiale quotidienne de protéines urinaires de plus de 2 g. Souvent, dans les cas de maladie plus avancée, l'excrétion quotidienne de protéines urinaires peut atteindre 5 g. -15 g.
L'insuffisance cardiaque se développe progressivement. Au moment où la plupart des patients atteints d'amylose AL présentent une pathologie cardiaque liée à l'amylose cliniquement apparente, des lésions myocardiques importantes ont déjà été observées. À la suite de l'expansion auriculaire, des tachyarythmies supraventriculaires peuvent survenir. La cardiomyopathie restrictive peut entraîner une hypotension orthostatique importante due à un remplissage ventriculaire limité, qui s'accompagne d'un dysfonctionnement autonome causé par des lésions du système nerveux périphérique.
Les saignements et le péristaltisme altéré sont les manifestations les plus courantes des dépôts amyloïdes dans le tractus gastro-intestinal. La satiété précoce, causée par une vidange gastrique retardée, est également un symptôme courant. Une croissance bactérienne excessive avec une malabsorption importante peut provoquer des diarrhées et entraîner des carences en vitamine B12, en acide folique et en carotène. L'hémorragie peut survenir dans n'importe quelle partie du tractus gastro-intestinal. bien que l'estomac et l'intestin grêle soient plus fréquemment touchés. Des dépôts d'amyloïde AL sont souvent observés dans le foie, bien que cela provoque rarement des symptômes.
L'atteinte du système nerveux périphérique, qui peut se développer des mois ou des années avant l'atteinte viscérale, survient chez jusqu'à 20 % des patients atteints d'amylose AL. Elle peut se manifester par une neuropathie sensorimotrice ou autonome, ou par une combinaison. Les paresthésies se développent d'abord dans les membres inférieurs et peuvent se propager de manière proximale avec le temps. L'atteinte des nerfs moteurs est rare, mais peut entraîner une déficience grave et entraîner un syndrome du pied tombant et des troubles de la marche. La neuropathie autonome est souvent observée chez les patients atteints d'amylose AL et entraîne une dysmotilité gastro-intestinale, une impuissance et une hypotension orthostatique.
Il existe deux manifestations pulmonaires prédominantes de l'amylose AL. Parfois, dans le parenchyme pulmonaire, l'amyloïde AL peut se présenter sous la forme d'une masse ressemblant à une tumeur, souvent avec une expansion concomitante des ganglions lymphatiques hilaires et péritrachéaux. Bien que ces masses puissent augmenter progressivement, elles ne mettent généralement pas la vie en danger.
Alternativement, une infiltration interstitielle diffuse du parenchyme pulmonaire peut se produire, ce qui provoque une raideur et une lésion pulmonaire restrictive. Rarement, l'amyloïde AL peut se déposer localement dans le larynx, la trachée, entraînant un enrouement et parfois une obstruction importante des voies respiratoires. Les anomalies hématologiques de l'amylose AL comprennent le purpura et la thrombose. L'infiltration amyloïde des vaisseaux sanguins provoque leur fragilité. Les ruptures des capillaires cutanés entraînent une extravasation de globules rouges et un purpura. Chez un patient atteint d'amylose AL, le purpura périorbitaire peut être causé par des activités relativement inoffensives, telles que se frotter les yeux ou incliner la tête pendant une longue période, entraînant des ecchymoses caractéristiques sous les yeux. Dans ce trouble, il existe une déficience en facteur X, qui serait due à l'absorption de ce facteur par d'importants dépôts d'amyloïde dans la rate, en raison de la perte de protéines dans le syndrome néphrotique. Ceci, associé à des perturbations du système plasminogène, entraîne une augmentation de la fréquence des thromboses veineuses.
Bien que l'amylose AL soit la forme la plus courante d'amylose, elle affecte rarement la peau, les muscles squelettiques et la langue, les modifications des tissus mous et des articulations sont rares. Le syndrome de la valve carpienne, souvent bilatéral, peut être causé par des dépôts amyloïdes dans le poignet entraînant une compression du nerf médian et peut être présent des années avant l'apparition d'une lésion systémique. L'infiltration amyloïde du muscle squelettique, impliquant généralement les tendons et les capsules des articulations de l'épaule, peut entraîner une pseudohypergrophie ("signe de l'épaulette") chez un patient en état de cachexie. Les dépôts amyloïdes dans les os, tels que le col fémoral, apparaissent sous forme de lumière kystique sur les radiographies et peuvent réduire la résistance osseuse, entraînant des fractures pathologiques. De rares cas de macroglossie ont été rapportés chez des patients atteints d'amylose AL. Une langue hypertrophiée et dure à la palpation peut causer des problèmes d'élocution et de déglutition et provoquer une sensation d'étouffement.
L'amylose AL résulte de l'expansion anormale et clonale des lymphocytes B. Cependant, l'expansion cellulaire monoclonale et la synthèse de chaînes légères ou lourdes sont des conditions nécessaires mais non suffisantes au développement de la maladie. L'amylose AL peut se développer avec la macroglobulinémie de Waldenström, le myélome multiple, la gammapathie monoclonale d'étiologie inconnue ou l'expansion bénigne des lymphocytes B. La quantité de protéine produite par ces clones ne semble pas avoir d'importance, puisque 10 à 20 % des patients atteints d'amylose AL n'ont pas de protéine monoclonale dans le sérum et l'urine. La structure primaire des chaînes légères est probablement d'une importance particulière dans le développement de cette maladie car les rapports normaux des chaînes légères sériques sont complètement altérés et les chaînes α. dans les dépôts d'amyloïde AL sont détectées beaucoup plus fréquemment que les chaînes k. Certains sous-types de chaînes L. sont plus sujets à la formation de dépôts fibrillaires que d'autres. De plus, les protéines fibrillaires AL-amyloïdes contiennent presque toujours un segment de chaîne légère variable (soit entièrement composé de celui-ci, soit le contenant sous forme de segment). Cependant, les causes des dommages sélectifs aux organes et les différents taux de progression de la maladie chez différents patients restent floues.
L'amylose AL est la maladie la plus grave parmi les amyloses, tandis que la durée de survie après le diagnostic ne dépasse pas 18 à 24 mois. L'apparition de la maladie avec un syndrome du canal carpien ou une neuropathie périphérique signifie souvent un meilleur pronostic que l'apparition d'une atteinte cardiaque. Une petite proportion de patients peut développer un myélome multiple après le diagnostic d'amylose AL, soulignant l'importance d'un suivi à long terme et de tests appropriés.
Le traitement de l'amylose AL vise à supprimer les clones de plasmocytes aberrants à l'aide de médicaments tels que le melphalan et la prednisone. Parfois, des médicaments de chimiothérapie tels que le cyclophosphamide ou le chlorambucil sont également utilisés. Les alcaloïdes de la pervenche et l'adriomycine doivent être utilisés avec beaucoup de prudence car ils peuvent être particulièrement toxiques chez les patients atteints de neuropathie ou de cardiomyopathie. Pour certains patients, le traitement de choix est le melphalan à haute dose avec greffe de cellules souches. Chez les patients atteints d'une maladie avancée, une dose intermédiaire de melphalan avec greffe de cellules souches peut être une alternative en raison d'une meilleure tolérance. Parmi les patients qui sont indiqués pour et subissant une greffe de moelle osseuse, l'espérance de vie moyenne atteint 40 mois, et chez les patients qui ne sont pas aptes à la greffe, elle est de 18 mois.
Amylose AA
L'amylose AA est la forme d'amylose systémique la plus répandue dans le monde. Tout stimulus inflammatoire peut provoquer une amylose AA. La cause la plus fréquente est la tuberculose; mais dans les pays industrialisés, les principales causes de l'amylose AA sont les maladies rhumatismales - polyarthrite rhumatoïde, spondyloarthrite et syndromes auto-inflammatoires. Les fibrilles d'amyloïde AA peuvent être détectées dans les biopsies de patients asymptomatiques, antérieures de plusieurs années à tout signe d'amylose systémique.
La manifestation la plus importante de l'amylose AA est une atteinte rénale, généralement présentée comme un syndrome néphrotique. Elle peut se développer 10 à 20 ans après le début de l'arthrite et peut survenir même après la disparition de la maladie inflammatoire primaire sous-jacente. Ainsi, l'amylose AA peut être confondue avec d'autres processus pathologiques impliquant le rein, comme la néphropathie induite par l'or. De plus, les déclencheurs inflammatoires aigus peuvent accélérer l'apparition de l'amylose AA systémique chez les patients qui ont déjà eu une maladie inflammatoire telle que la tuberculose ou d'autres infections chroniques. C'est pourquoi les patients atteints d'une nouvelle tuberculose active peuvent développer un syndrome néphrotique en quelques semaines, peut-être parce que des foyers préexistants de dépôts amyloïdes localisés peuvent accélérer la progression de l'amylose AA systémique.
Les patients atteints d'amylose AA peuvent présenter des saignements gastro-intestinaux. Le dépôt de la protéine AA dans la paroi du vaisseau sanguin entraîne une diminution de l'extensibilité et une augmentation de la fragilité, avec parfois des ruptures de vaisseaux et des saignements. Bien que décrites dans la littérature, des lésions importantes du cœur, des nerfs, des muscles squelettiques ou de la langue sont très rares dans l'amylose AA. Il est important d'exclure la présence d'amylose AA chez les patients atteints d'un syndrome néphrotique sévère, même chez ceux qui n'ont pas d'antécédents de maladie inflammatoire ou infectieuse. Ce schéma est observé chez les patients atteints de fièvre méditerranéenne familiale qui présentent des élévations subcliniques du SAA et d'autres protéines de phase aiguë, mais aucun autre symptôme. En fin de compte, la maladie chez ces patients peut évoluer vers une amylose systémique. Étant donné que bon nombre de ces patients vivaient dans des pays en développement, il est possible que des facteurs environnementaux, tels que des infections endémiques qui provoquent une inflammation chronique, contribuent à ce schéma de maladie, augmentant ainsi le risque de développer une amylose AA.
Le traitement vise à contrôler le processus inflammatoire sous-jacent. L'évolution clinique de l'amylose AA est plus favorable lorsque la concentration en SAA reste inférieure à 10 mg/l. Dans une forme plus grave de la maladie chez les patients atteints d'amylose AA, la fonction rénale est efficacement restaurée par la transplantation rénale. Cependant, si le processus inflammatoire sous-jacent n'est pas supprimé, l'amyloïde AA peut également se déposer dans le rein transplanté.
Amylose ATTR
Les amyloses héréditaires sont causées par diverses protéines non apparentées. Ces syndromes sont hérités de manière autosomique dominante. La mutation génétique est présente à la naissance, mais les symptômes cliniques de la maladie n'apparaissent généralement qu'à la fin de la troisième décennie de la vie. Ces syndromes ont des manifestations cliniques similaires et s'accompagnent du développement d'une cardiomyopathie, d'une néphropathie et d'une polyneuropathie. Cependant, chaque protéine amyloïdogène doit être considérée comme provoquant une maladie indépendante avec des caractéristiques cliniques uniques. La grande majorité des amyloses héréditaires sont causées par le dépôt de variants de la transthyrétine (TTR), pour lesquels plus d'une centaine de mutations ont été identifiées. Le TTR est également connu sous le nom de pré-albumine car il se déplace plus rapidement en électrophorèse sur gel que l'albumine. La transthyrétine est une protéine plasmatique qui transporte environ 20 % de la thyroxine dans le plasma, ainsi que de la vitamine A associée à la protéine liant le rétinol. Le TTR est synthétisé dans le foie sous la forme d'un seul polypeptide et forme un tétramère dans le plasma, composé de quatre monomères identiques. La protéine de type sauvage a une structure pliée prononcée ; le remplacement d'un seul acide aminé provoque son agrégation et la formation de fibrilles.
Toutes les amyloses associées au TTR ne sont pas dues à des mutations du TTR. Des fragments de TTR de type sauvage peuvent former des fibrilles amyloïdes qui se déposent dans le cœur, provoquant une amylose cardiaque sénile. Cette maladie non héréditaire touche environ 25 % des personnes de plus de 80 ans.
La plupart des amyloses associées au TTR se présentent initialement comme une neuropathie périphérique. Il s'agit souvent d'une neuropathie sensorimotrice impliquant les membres inférieurs distaux qui évolue vers les membres proximaux. Dans 20% des cas, la manifestation initiale peut être un syndrome du canal carpien à la suite d'une compression du nerf médian par des dépôts amyloïdes d'ATTR. La neuropathie autonome peut provoquer des symptômes gastro-intestinaux tels que l'alternance de constipation et de diarrhée, ou des symptômes génito-urinaires tels que l'incontinence ou l'impuissance.
Bien que les dommages au système nerveux périphérique soient associés à une altération significative, les principales causes de décès chez les patients atteints d'amylose ATTR sont la cardiomyopathie et les maladies rénales. La plupart des décès (60 %) sont dus à une cardiomyopathie, tandis que les lésions rénales ne causent que 5 à 7 % des décès et que des dépôts amyloïdes vitreux sont observés chez 20 % des patients atteints d'amylose ATTR. On pense qu'ils résultent de l'accumulation de TTR, qui est sécrétée par le plexus choroïde et forme des fibrilles amyloïdes qui s'accumulent dans le vitré.
L'amylose ATTR est diagnostiquée en utilisant des méthodes génétiques pour détecter les mutations du TTR, la plupart des mutations de l'ATTR se produisant dans les exons 2-4. La réalisation d'une réaction en chaîne par polymérase pour détecter les polymorphismes des fragments de restriction est devenue une méthode courante pour diagnostiquer la maladie et identifier les porteurs du gène mutant parmi les membres de sa famille.
L'amylose ATTR est traitée par transplantation du foie ou d'autres organes malades. La transplantation hépatique entraîne une synthèse de TTR de type sauvage (normale), avec disparition rapide du variant transthyrétine de la circulation. Les patients atteints d'amylose ATTR avec des lésions rénales importantes subissent une transplantation combinée foie / rein. Il est important que les patients atteints d'amylose ATTR soient traités avant le développement d'une malnutrition sévère ou d'une cardiomyopathie, car la survie du greffon diminue rapidement lorsque de tels changements se développent. Le dépôt d'amyloïde peut continuer même après la transplantation d'organe, peut-être en raison de la présence des plus grands dépôts de protéines anormales, qui servent de noyau pour le dépôt ultérieur de protéines normales. Pour cette raison, les patients présentant des manifestations antérieures d'amylose ATTR peuvent nécessiter une nouvelle transplantation d'organe.
Amylose Ap2M
Les dépôts d'Ap2M-amyloïde sont principalement localisés dans les tissus du système musculo-squelettique. La présence de douleurs dans l'articulation de l'épaule, d'un syndrome du canal carpien et de contractures persistantes en flexion des doigts chez un patient sous hémodialyse au long cours suggère une amylose Ap2M ou liée à la dialyse). Les signes et symptômes de l'amylose Ap2M sont parfois observés chez les insuffisants rénaux chroniques qui n'ont pas encore subi de dialyse.
Les lésions squelettiques axiales, qui surviennent chez 10 % des patients sous hémodialyse à long terme, se manifestent par une spondylarthropathie destructrice, dont les caractéristiques radiographiques incluent une diminution de la hauteur des disques intervertébraux et une érosion des plateaux vertébraux sans formation prononcée d'ostéophytes. Le plus souvent, la partie inférieure de la colonne cervicale est touchée ; cependant, des changements similaires peuvent également être observés dans la colonne thoracique et lombaire. Des dépôts kystiques d'Ap2M-amyloïde ont été trouvés dans le processus odontoïde et les corps des vertèbres cervicales supérieures, ainsi que des masses d'Ap2M-amyloïde dans les tissus mous parodontoïdes, appelées pseudotumeurs. Bien que les troubles neurologiques soient rares, une myélopathie sévère survient en raison de dépôts d'amyloïde Ap2M dans la colonne cervicale et lombaire, en particulier chez les patients qui ont subi une hémodialyse pendant 20 ans ou plus.
Des lésions osseuses kystiques peuvent se développer dans les os du squelette périphérique des patients qui ont subi une hémodialyse à long terme. Les kystes amyloïdes sous-chondraux se trouvent généralement dans les os du poignet, mais peuvent également apparaître dans l'acétabulum et les os longs tels que la tête ou le cou du fémur, la tête de l'humérus, le radius distal et le tibia supérieur. Contrairement aux tumeurs brunes de l'hyperparathyroïdie, ces kystes osseux se produisent généralement dans les tissus adjacents aux articulations et augmentent en taille et en nombre avec le temps. Des fractures pathologiques, en particulier du col fémoral, peuvent survenir sur des os fragilisés par des dépôts amyloïdes.
Les patients dialysés depuis plus de 10 ans présentent des dépôts viscéraux d'amyloïde Ap2M. Bien que des complications dans le tractus gastro-intestinal et le système cardiovasculaire aient été décrites, les dépôts viscéraux d'amyloïde Ap2M ne provoquent généralement pas de symptômes.
Les théories modernes de la pathogenèse de l'amylose Ap2M impliquent la participation du produit final de glycosylation améliorée (AGE) à la modification des protéines, ce qui contribue à leur résistance à la protéolyse, augmente leur affinité pour le collagène et la capacité à stimuler la sécrétion de protéines pro-inflammatoires. cytokines telles que TNF-α, IL-6 par des leucocytes mononucléaires activés. Les protéines modifiées par les AGE sont mal excrétées par dialyse. Ainsi, les patients sous dialyse ont des concentrations élevées de ces protéines modifiées par rapport aux individus ayant une fonction rénale normale ou des allogreffes rénales fonctionnelles. Les patients présentant des symptômes et des dépôts massifs d'amyloïde Ap2M peuvent nécessiter une intervention chirurgicale. Au cours de la dernière décennie, l'utilisation de nouvelles membranes plus perméables en hémodialyse a probablement retardé l'apparition du syndrome du canal carpien et des kystes osseux, et réduit l'incidence de l'amylose Ap2M. Les dépôts d'amyloïde Ap2M ne sont pas progressifs et peuvent régresser chez les patients qui ont eu une greffe de rein réussie. Les patients atteints d'amylose Ap2M qui ont eu une greffe de rein réussie connaissent une réduction marquée des douleurs et des raideurs articulaires. Ainsi, une transplantation rénale précoce chez les candidats éligibles avant que des dépôts amyloïdes AP2M importants ne se développent peut être la mesure préventive la plus efficace disponible pour cette maladie.
Amylose des organes internes
Les formes localisées d'amylose peuvent affecter divers organes et systèmes, notamment les yeux, les voies génito-urinaires, le système endocrinien et les voies respiratoires. À l'exception de la maladie d'Alzheimer, ces types d'amylose sont rares et difficiles à diagnostiquer. Les principes physiopathologiques régissant la manifestation de la maladie dans les formes localisées sont similaires à ceux observés pour les formes systémiques. Les formes les plus courantes d'amylose localisée concernent les voies génito-urinaires et respiratoires.
Amylose génito-urinaire
L'amylose urogénitale localisée peut impliquer l'ensemble du tractus, mais le plus souvent la vessie et l'urètre sont impliqués, provoquant une hématurie ou des signes d'obstruction. La protéine amyloïde est souvent représentée par des chaînes légères ou lourdes d'immunoglobulines. La détection de dépôts amyloïdes locaux peut induire une recherche invalidante de maladie systémique, souvent avec des résultats négatifs. Cependant, l'amylose localisée se résout généralement spontanément et ne laisse pas présager un mauvais pronostic. Le traitement consiste en l'excision des dépôts amyloïdes localisés.
Amylose des poumons
Dans les voies respiratoires, le dépôt d'AL amyloïde provoque souvent des formes localisées de la maladie. Les voies respiratoires sont concernées par trois formes d'amylose localisée : l'amylose trachéobronchique. qui représente la moitié des cas ; l'amylose parenchymateuse nodulaire, qui survient dans environ 45 % des cas ; et l'amylose parenchymateuse diffuse, qui représente environ 5 % des cas. Dans l'amylose trachéobronchique, il existe une atteinte localisée ou diffuse de l'arbre trachéobronchique avec dépôt sous-muqueux d'amyloïde. La tomodensitométrie (TDM) révèle des nodules ou des plaques d'amyloïde, parfois avec calcification ou épaississement annulaire de la trachée, des bronches principales, des bronches lobaires ou segmentaires avec rétrécissement de la lumière. Dans l'amylose parenchymateuse nodulaire, la TDM montre des nodules à bords nets et lobulaires, localisés en périphérie et sous-pleuralement. Les nodules varient en taille d'un micronodule à 15 cm de diamètre; dans la moitié des cas, une calcification est observée. L'amylose septale parenchymateuse ou alvéolaire diffuse présente des dépôts amyloïdes étendus impliquant de petits vaisseaux et du tissu interstitiel parenchymateux ; de petits nodules amyloïdes multifocaux peuvent également être présents. La tomodensitométrie haute résolution montre des opacités rétiniennes anormales, un épaississement septal interlobulaire, de petits nodules (2 à 4 mm de diamètre) et des opacités regroupées confluentes principalement dans les zones sous-pleurales. Ce schéma d'amylose localisée est parfois impossible à distinguer de l'amylose systémique. Les patients atteints de cette forme d'amylose pulmonaire parenchymateuse diffuse sont plus susceptibles de mourir d'une insuffisance respiratoire que les patients atteints d'amylose parenchymateuse trachéobronchique ou nodulaire.
Le dépôt amyloïde localisé limité aux voies respiratoires peut être réséqué pour traiter cette forme d'amylose localisée. D'autres types d'amyloïde peuvent également se déposer dans les voies respiratoires, mais cela est rare et n'entraîne généralement pas de pathologie importante.
Méthodes de diagnostic de l'amylose
La scintigraphie à l'amyloïde P sérique permet d'identifier la distribution systémique des dépôts amyloïdes.Les images en série montrent la progression et la régression des dépôts amyloïdes. Cependant, cette technique est limitée car les patients sont exposés à la protéine allogénique radioactive et n'est disponible que dans des centres spécialisés.
La seule technique d'imagerie largement disponible qui fournit des informations spécifiques au diagnostic de l'amylose systémique est l'échocardiographie. Les caractéristiques échocardiographiques spécifiques de l'amylose comprennent la dilatation auriculaire, la contraction ventriculaire gauche, l'épaississement du septum interventriculaire et interauriculaire et l'augmentation de l'échogénicité myocardique. Dans un stade ultérieur, des changements restrictifs plus prononcés sont notés. Malheureusement, l'espérance de vie moyenne après l'apparition des signes échocardiographiques d'amylose n'est que de 6 mois. De plus, l'échocardiographie ne révèle pas de régression de l'amylose même après un traitement réussi.
L'imagerie par résonance magnétique (IRM) du cœur est un domaine de recherche en plein essor qui complète l'échocardiographie dans le diagnostic de l'amylose cardiaque. L'IRM cardiaque avec contraste de gadolinium a une haute résolution (environ 2 mm) et fournit un contraste tissulaire, permettant de différencier la zone affectée du myocarde normal. Chez les patients atteints de maladie cardiaque amyloïde, l'IRM cardiaque démontre une amélioration qualitative du contraste total et sous-endocardique après l'administration intraveineuse de gadolinium Bien qu'il n'y ait pas de preuve typique d'amylose cardiaque par IRM, des études futures pourraient déterminer une combinaison de techniques non invasives qui pourraient être utilisées dans la sélection des patients pour certaines biopsies plus invasives de l'endomyocarde, ainsi que pour suivre l'évolution naturelle de l'amylose cardiaque.
Puisqu'il n'y a pas de signes spécifiques à l'amylose systémique, l'imagerie doit être utilisée en complément de l'examen clinique et des tests de laboratoire appropriés pour évaluer les patients présentant des symptômes caractéristiques. Bien que le tractus gastro-intestinal soit presque toujours impliqué dans l'amylose systémique, les signes radiographiques d'amylose gastro-intestinale sont rares. L'ischémie et le dépôt d'amyloïde dans les vaisseaux peuvent provoquer un épaississement symétrique des plis muqueux, qui sont détectés au scanner.
Ou les tomodensitogrammes aident à détecter l'hypertrophie rénale dans les premiers stades de l'amylose. L'échographie montre généralement une augmentation diffuse de l'échogénicité du parenchyme rénal avec préservation du contraste cortico-médullaire car l'architecture de la couche corticale aux premiers stades de la maladie reste macroscopiquement normale. La progression de la maladie peut s'accompagner d'une diminution du rein et d'un amincissement important de la couche corticale.
Si une amylose est suspectée, le diagnostic est confirmé par biopsie : la microscopie du matériel en lumière polarisée révèle une biréfringence vert clair caractéristique et, à l'aide d'études immunohistochimiques, le type de protéine amyloïde. Une biopsie peut être prélevée sur un organe affecté ou non. Cette dernière approche est généralement préférée en raison du risque élevé de complications et d'inconfort associés à la biopsie viscérale. L'une des trois méthodes est couramment utilisée pour diagnostiquer l'amylose : la biopsie du tractus gastro-intestinal (rectal ou gastroduodénal), l'aspiration de la graisse abdominale sous-cutanée et la biopsie de la glande salivaire mineure.
La biopsie rectale réalisée par sigmoïdoscopie ou sigmoïdoscopie est la biopsie préférée du tractus gastro-intestinal en raison de l'accessibilité de ce site. La biopsie doit inclure les vaisseaux sanguins sous-muqueux, qui sont plus susceptibles de contenir des dépôts amyloïdes que ceux de la muqueuse ou des couches musculaires. Bien que les résultats les plus fiables puissent être obtenus à partir d'une biopsie rectale, une biopsie de l'estomac ou du duodénum peut également diagnostiquer l'amylose si l'échantillon de tissu contient des vaisseaux sanguins de la taille appropriée.
L'aspiration de la graisse abdominale a été réalisée pour la première fois après qu'il a été observé que les échantillons d'autopsie de patients atteints d'amylose contiennent souvent des dépôts amyloïdes autour des adipocytes ; la plus forte densité de dépôts amyloïdes a été observée dans les tissus adipeux du cuir chevelu et de la paroi abdominale. La sensibilité de la ponction de graisse abdominale varie entre 55 et 75 %, mais est similaire à celle d'une biopsie rectale. Cette technique est utile pour diagnostiquer l'amylose AA, AL et ATTR ; cependant, en raison de la distribution limitée des dépôts d'amyloïde Ap2M dans les organes, l'aspiration de la graisse abdominale peut ne pas être une méthode fiable pour diagnostiquer l'amylose Ap2M.
Avec une biopsie de la glande salivaire mineure, des glandes salivaires supplémentaires de la membrane muqueuse de la lèvre sont prélevées. Auparavant, la biopsie gingivale était utilisée pour détecter les dépôts amyloïdes, mais la sensibilité de cette méthode s'est avérée faible. Dans les amyloses AA, ATTR et AL, la sensibilité d'une biopsie mineure des glandes salivaires est comparable à celle d'une biopsie rectale ou d'une aspiration de graisse abdominale.
Si la suspicion d'amylose est importante et qu'aucune des méthodes ci-dessus ne donne de résultats positifs, il est nécessaire d'effectuer une biopsie de l'organe affecté. Lorsque les reins sont impliqués, une biopsie rénale fournit généralement des informations diagnostiques. Dans les amyloses ATTR et AL, le cœur et la moelle osseuse sont touchés. Une biopsie de ces organes est donc nécessaire pour confirmer le diagnostic. Bien que le nerf sural puisse être impliqué, il est moins souhaitable de le biopsier car la procédure est généralement douloureuse, la plaie de biopsie guérit lentement et il peut y avoir une altération sensorielle résiduelle. De plus, la distribution inégale des dépôts amyloïdes fait de la biopsie du nerf sural une procédure moins sensible que la biopsie d'autres organes affectés.
Lors du diagnostic d'amylose, trois points revêtent une importance particulière ::
- La probabilité pré-test de détecter l'amyloïde dans une biopsie est déterminée par les manifestations cliniques de la maladie. Pour déterminer la probabilité du prétest, il est important de tenir compte des antécédents (y compris des antécédents familiaux complets), d'un examen clinique complet et d'une évaluation de laboratoire, qui comprend une électrophorèse des protéines sériques et urinaires, et une analyse d'urine pour évaluer le degré de protéinurie.
- L'immunohistochimie doit toujours être effectuée sur des échantillons de tissus qui sont évalués pour les dépôts amyloïdes afin d'identifier la protéine amyloïde spécifique. Occasionnellement, un patient atteint d'une maladie inflammatoire peut développer une amylose AL, ou un patient porteur d'une protéine monoclonale sérique peut développer une amylose AA. Le traitement de ces maladies étant très différent, il est impératif d'établir un diagnostic précis.
- Des dépôts d'amyloïde AA dans la graisse abdominale sont souvent observés dans les maladies inflammatoires, telles que la polyarthrite rhumatoïde ou la spondylarthrite ankylosante. Cependant, même après un suivi à long terme, la plupart de ces patients ne présentent aucun signe de dysfonctionnement organique. Ainsi, toutes les personnes présentant des dépôts d'amyloïde AA ne sont pas atteintes d'amylose AA; les résultats de la biopsie doivent être interprétés avec prudence.
L'amylose est une maladie difficile à diagnostiquer et à traiter et souvent de mauvais pronostic. Il existe de nombreux types différents de la maladie, mais les plus connus sont l'amylose primaire et secondaire. Lors de la détermination de la pathologie à un stade précoce du développement, une réponse positive au traitement est possible.
L'amylose est un trouble du métabolisme des protéines, qui s'accompagne du dépôt dans divers tissus et organes d'une protéine spécifique appelée amyloïde. Les manifestations cliniques dépendent du type de maladie et sont principalement définies comme des variables.
Le groupe de troubles systémiques appelés "amylose" comprend environ 30 types différents, différant les uns des autres par la spécificité des troubles protéiques. Les quatre plus connues sont l'amylose AL, l'amylose AA, l'amylose AF et l'amylose AH.
Le diagnostic peut être posé par la détection de protéines dans les urines ou par la présence d'anomalies dans les organes internes sans aucune raison. La maladie est confirmée par une biopsie tissulaire. La thérapie vise principalement à réduire la concentration de la protéine anormale ou la cause de la maladie.
Vidéo : Qu'est-ce que l'amylose, pourquoi est-elle dangereuse, comment y faire face ?
Description
L'amylose est un trouble systémique classé en plusieurs types classés comme primaire, secondaire ou familial (héréditaire).
- Amylose primaire (AL ) est le type le plus courant d'amylose systémique. La LA est le résultat d'une anomalie (dyscrasie) des plasmocytes (un type de globules blancs) dans la moelle osseuse et est étroitement associée au myélome multiple.
- Secondaire (AA) l'amylose dans son développement repose sur la détermination de la protéine inflammatoire amyloïde sérique. Elle est souvent associée à une maladie inflammatoire chronique telle que les maladies rhumatismales, la fièvre méditerranéenne familiale, la maladie intestinale inflammatoire chronique, la tuberculose ou l'empyème.
- Amylose familiale est un type rare d'amylose causée par un gène anormal. Plusieurs gènes anormaux peuvent causer la maladie, mais le type le plus courant d'amylose héréditaire est appelé ATTP, causé par des mutations de la transthyrétine (TTR).
- Amylose sénile , dans laquelle la protéine anormale est dérivée de la transthyrétine dinate (normale), est une maladie à évolution lente affectant le muscle cardiaque chez les personnes âgées.
Les dépôts amyloïdes peuvent parfois se produire isolément sans signe de maladie systémique. Par exemple, cela inclut une lésion vésicale unique ou une amylose trachéale, les types les plus courants d'amylose isolée.
L'amylose à bêta2-microglobuline associée à la dialyse est un type d'amylose systémique qui survient chez les personnes qui ont eu de longues périodes pour éliminer les toxines accumulées ou les déchets du sang par filtration mécanique. Cette forme d'amylose, également connue sous le nom d'ABM2 (protéine bêta-2m associée à l'amyloïde), se produit en raison de l'agrégation de la bêta2-microglobuline, un type de protéine amyloïde qui est éliminée dans un rein fonctionnant normalement. L'amylose à bêta2-microglobuline associée à la dialyse survient également chez les patients présentant une insuffisance rénale tardive au cours de la maladie. Cependant, la maladie ne se manifeste pas chez les personnes ayant une fonction rénale normale ou modérément réduite ou chez les patients après une transplantation rénale.
panneaux
L'amylose est généralement une maladie multisystémique entraînant un large éventail de manifestations cliniques. Ainsi, le patient peut être vu par plusieurs spécialistes, le plus souvent par un néphrologue, un cardiologue ou un neurologue. La plupart des patients ont une atteinte de plusieurs organes, il n'est donc pas rare qu'une combinaison des éléments suivants soit présente et, si elle est présente, une amylose doit être suspectée :
- reins- le plus souvent atteint dans l'AL, l'AA et certaines formes héréditaires rares d'amylose, mais survient rarement dans les formes familiales causées par des mutations de la transthyrétine. L'excès de protéines dans l'urine (protéinurie) est une manifestation courante de lésions rénales et est souvent grave, entraînant un syndrome néphrotique. L'amyloïde provoque un excès d'urée et d'autres déchets azotés dans le sang (azotémie progressive) et est la première manifestation d'une maladie rénale. Une accumulation anormale de liquide (œdème), en particulier dans les jambes et l'abdomen, en l'absence d'insuffisance cardiaque, est un signe de syndrome néphrotique. De plus, la présence d'un excès de cholestérol dans le sang (hypercholestérolémie) peut atteindre un degré de gravité profond. Les reins de l'amylose diminuent de taille, pâlissent et deviennent plus lourds, mais de gros reins sont généralement observés dans l'amylose. De plus, l'hypertension artérielle (hypertension) et la thrombose du tissu rénal peuvent être déterminées. L'amyloïde peut s'accumuler dans d'autres parties du système génito-urinaire, comme la vessie ou les uretères.
Vidéo : Elena Malysheva. Amylose rénale
- Cœur. L'amylose affecte souvent le tissu cardiaque. L'infiltration amyloïde du cœur entraîne un épaississement de la paroi ventriculaire et le développement d'une insuffisance cardiaque. L'insuffisance cardiaque congestive rapidement progressive avec des parois ventriculaires épaisses est l'image classique de l'amylose cardiaque. Le myocarde est invariablement affecté dans l'amylose sénile, l'amylose TTP, et n'est presque jamais impliqué dans l'amylose secondaire. Les symptômes courants de l'amylose cardiaque comprennent :
- une hypertrophie cardiaque (cardiomégalie);
- rythme cardiaque irrégulier (arythmies);
- murmures dans le cœur;
- anomalies cardiaques observées à l'électrocardiographie (p. ex., faible tension de forme d'onde).
L'insuffisance cardiaque congestive est la complication la plus fréquente de l'amylose. Des dépôts amyloïdes nodulaires peuvent être présents sur la membrane qui entoure le cœur (péricarde) et sur la couche interne des cavités ou valves cardiaques (endocarde).
- Système nerveux. Bien que moins fréquente que l'insuffisance rénale ou cardiaque, la neuropathie peut être un problème important dans l'amylose. Assez fréquent dans l'amylose AL. La neuropathie est souvent indolore et de nature sensorimotrice, bien que la douleur neuropathique puisse parfois être importante. Les symptômes peuvent inclure :
- neuropathie sensorielle avec engourdissement et picotements dans les jambes qui progressent le long des jambes et finissent par se propager aux membres supérieurs ;
- neuropathie motrice avec perte de mouvement, commençant dans les jambes et s'étendant vers le haut.
Le syndrome du canal carpien n'est généralement pas dû à une atteinte nerveuse directe, mais plutôt à une infiltration des tissus mous qui contribue à la compression nerveuse. Dans l'amylose familiale, la neuropathie périphérique s'accompagne souvent d'une neuropathie autonome caractérisée par une diarrhée et une diminution de la transpiration (hypohidrose), une chute brutale de la pression artérielle lorsque le patient se lève (hypotension orthostatique) et, chez l'homme, une dysfonction érectile.
L'hypotension orthostatique peut être profonde et entraîner des épisodes répétés d'évanouissements (syncopes). L'amylose systémique n'est pas associée au système nerveux central ni à la maladie d'Alzheimer.
- Organes digestifs. L'amylose peut affecter le foie et la rate. Les dommages au dernier organe augmentent le risque de rupture traumatique. L'atteinte hépatique est fréquente dans l'amylose AL. Il se produit également dans l'amylose AA mais n'est pas observé dans l'amylose TTR familiale. Dans la plupart des cas, l'atteinte hépatique est asymptomatique. Les signes les plus notables sont
- hypertrophie du foie (hépatomégalie);
- hypertrophie de la rate (splénomégalie).
En règle générale, les lésions amyloïdes du foie s'accompagnent d'une augmentation des enzymes (en particulier de la phosphatase alcaline) et d'autres fonctions organiques, souvent détectées à un stade précoce. En règle générale, la fonction hépatique n'affecte pas de manière significative l'évolution de la maladie aux stades ultérieurs. Une augmentation de la bilirubine est un signe défavorable et peut laisser présager une insuffisance hépatique.
L'accumulation d'amyloïde dans le tractus gastro-intestinal peut entraîner un manque de mouvement (motilité) de l'œsophage et de l'intestin grêle et du gros intestin. Vous pouvez également rencontrer :
- malabsorption;
- ulcération;
- saignement;
- faible activité de l'estomac;
- pseudo-obstruction du tractus gastro-intestinal;
- perte de protéines;
- diarrhée.
- Cuir souvent affecté dans l'amylose primaire. Le purpura périorbitaire est le résultat d'une fragilité capillaire et peut apparaître après avoir toussé, éternué ou forcé avec une selle. Il n'est pas rare que des lésions violettes se produisent après quelque chose d'aussi simple que de se frotter les paupières. L'infiltration des tissus mous provoque une macroglossie et un enrouement, bien que l'examen des cordes vocales puisse ne pas révéler d'anomalies. Les lésions cutanées sont parfois très visibles ou si mineures qu'elles nécessitent l'utilisation d'un microscope pour les diagnostiquer.
Des lésions papuleuses cireuses peuvent apparaître sur le visage et le cou. On les trouve aussi souvent sous les aisselles (aisselle), près de l'anus, et dans l'aine. Les autres zones susceptibles d'être touchées sont les zones muqueuses, le conduit auditif et la langue. Peuvent également être présents :
- gonflement;
- saignement sous la peau (purpura);
- perte de cheveux (alopécie);
- inflammation de la langue (glossite);
- bouche sèche (xérostomie).
- Système respiratoire. Les problèmes respiratoires associés à l'amylose se développent souvent en parallèle avec des problèmes cardiaques. Dans une forme localisée d'amylose, les voies respiratoires peuvent être bloquées par des dépôts amyloïdes dans les sinus, le larynx, la trachée et l'arbre bronchique. L'accumulation de liquide dans l'espace pleural (épanchement pleural) est assez fréquente chez les patients atteints d'insuffisance cardiaque congestive due à l'amylose. De grands épanchements pleuraux récurrents disproportionnés par rapport au degré d'insuffisance cardiaque indiquent une amylose pleurale.
L'arthropathie survient dans l'amylose en raison de l'accumulation de dépôts amyloïdes dans les membranes synoviales. Cela se produit dans l'amylose AL et occasionnellement dans l'amylose de dialyse. Le cartilage articulaire ou la membrane synoviale et le liquide peuvent également être impliqués dans le processus pathologique. Les symptômes sont similaires à ceux de la polyarthrite rhumatoïde.
Les dépôts amyloïdes dans les tissus musculaires peuvent provoquer une faiblesse musculaire et des modifications musculaires (pseudomyopathies). Les symptômes de l'amylose se manifestent également souvent par des saignements. Cela peut être le résultat d'une déficience de certains facteurs de coagulation ou de petits dépôts amyloïdes dans les vaisseaux sanguins de la peau.
causes
L'amylose est causée par des protéines anormales qui favorisent la formation de fibrilles dans un ou plusieurs organes, systèmes ou tissus mous. Ces accumulations de protéines sont appelées dépôts amyloïdes, qui peuvent entraîner une détérioration progressive et un dysfonctionnement complet de l'organe affecté. Normalement, les protéines sont décomposées à peu près au même rythme qu'elles sont fabriquées, mais les dépôts amyloïdes exceptionnellement stables se déposent plus rapidement qu'ils ne sont décomposés.
Cause de l'amylose primaire (AL) est généralement une dysclasie des plasmocytes, une anomalie acquise des plasmocytes dans la moelle osseuse avec une production anormale de protéines. Habituellement, une quantité excessive de protéines se forme, qui s'accumule dans les tissus du corps sous la forme de dépôts amyloïdes.
Amylose secondaire (AA ) est causée par un processus inflammatoire qui fait partie de la maladie sous-jacente. Environ 50 % des personnes atteintes d'amylose secondaire souffrent de polyarthrite rhumatoïde comme affection sous-jacente.
L'amylose familiale est causée anomalies dans un gène pour l'une de plusieurs protéines spécifiques. La forme la plus courante d'amylose héréditaire est causée par une anomalie (mutation) du gène de la transthyrétine. Plus de 100 mutations différentes de la transthyrétine ont été rapportées et la mutation la plus courante a été nommée V30M. Les mutations du TTR sont principalement associées à l'amylose, qui affecte divers systèmes d'organes. Rarement, les mutations des gènes protéiques qui causent l'amylose sont la chaîne A du fibrinogène afilique, les apolipoprotéines A1 et A2, la gelsoline et la cystatine C.
Toutes les amyloses héréditaires sont associées à un mode de transmission autosomique dominant. La plupart des maladies génétiques sont déterminées par le statut de deux copies du gène reçues du père et une de la mère. Les troubles génétiques dominants surviennent lorsqu'il suffit d'une seule copie d'un gène anormal pour provoquer une maladie particulière. Le gène anormal peut être hérité de l'un ou l'autre parent ou peut être le résultat d'une nouvelle mutation (changement de gène) chez l'individu affecté.
Le risque de transmettre le gène anormal d'un parent affecté à la progéniture est de 50 % à chaque grossesse. Le risque est le même pour les hommes et les femmes. Cependant, toutes les personnes qui reçoivent le gène ne développeront pas nécessairement une amylose.
La cause exacte de l'amylose à bêta2-microglobuline associée à la dialyse n'est pas entièrement comprise. Un rein fonctionnant normalement peut se débarrasser de la protéine amyloïde, la bêta2-microglobuline. Chez certains patients sous dialyse de longue durée ou sous dialyse péritonéale continue ambulatoire, l'incapacité des reins à fonctionner normalement entraîne une rétention et une accumulation anormales de la protéine bêta2-microglobuline. Certaines personnes atteintes d'insuffisance rénale terminale développent également cette forme d'amylose.
Diagnostique
Le diagnostic d'amylose est suspecté après une histoire détaillée et une présentation clinique, mais une biopsie du muscle, de l'os ou du tissu adipeux est nécessaire pour confirmer la présence d'amyloïde.
Si la maladie est suspectée sur des bases cliniques, une biopsie de l'organe impliqué donnera le résultat le plus fiable. Le matériel de biopsie est examiné au microscope et coloré avec un colorant appelé rouge Congo. Lorsque le diagnostic d'amylose est posé par biopsie tissulaire, un examen approfondi du patient est effectué pour déterminer quels organes sont touchés.
Une fois que l'amyloïde est déterminée par biopsie tissulaire, le type de maladie doit être déterminé. Dans l'amylose AL, une dyscrasie des plasmocytes se manifeste, retrouvée dans 98% des cas. Dans 2 % des cas, le lymphome à cellules B est identifié comme la cause de la LA.
Les tests spécifiques utilisés pour établir un diagnostic de type amylose AL sont les suivants :
- électrophorèse des protéines du sang et de l'urine;
- biopsie de la moelle osseuse avec coloration immunochimique des plasmocytes ;
- analyse acellulaire de la chaîne légère.
Le diagnostic d'amylose AL est confirmé par la présence d'un purpura périorbitaire, qui résulte d'une fragilité capillaire, ou d'une macroglossie (langue hypertrophiée).

Le diagnostic d'amylose TTR héréditaire peut être confirmé en réalisant tests génétiques moléculaires , qui détecte les mutations du gène TTR à partir d'un échantillon de sang. En l'absence de mutations de la transthyrétine, de très rares formes d'amyloïde familiale peuvent être présentes.
Si le patient est une personne âgée atteinte d'insuffisance cardiaque cliniquement isolée, le diagnostic le plus probable est l'amylose systémique sénile. Il s'agit d'une condition dans laquelle la transthyrétine dystrophique (normale) se dépose dans le cœur.
Immunomarquage spécifique e(par exemple, la microscopie électronique immunopharyngée) est disponible dans des centres spécialisés et constitue un test très spécifique pour déterminer le type d'amyloïde.
Dans les cas difficiles de diagnostic spectrométrie de masse capable de déterminer avec précision la structure moléculaire des dépôts amyloïdes, une technique de plus en plus utilisée.
La méthode appelée balayage de sérum amyloïde P marqué radioactif , est disponible dans plusieurs centres européens spécialisés dans l'amylose. Ce test est utilisé pour surveiller l'accumulation de dépôts amyloïdes.
Chez les personnes sous dialyse à long terme ou souffrant d'insuffisance rénale terminale, des tests de laboratoire peuvent être effectués pour analyser des échantillons de sang ou d'urine afin de vérifier les niveaux élevés de la protéine B2M.
Thérapie standard
La stratégie de traitement dépend du type d'amylose et de l'état clinique du patient. Dans l'amylose AL, la cause est un globule blanc anormal (généralement un plasmocyte), et donc le pilier du traitement pour ce type d'amylose est la chimiothérapie pour éradiquer ces cellules.
Depuis de nombreuses années, le melphalan et la dexaméthasone sont utilisés par voie orale ou intraveineuse, souvent associés à un support de cellules souches autologues.
Les deux médicaments sont tout aussi efficaces, mais le traitement et les effets secondaires sont différents. Le melphalan à haute dose avec support de cellules souches est un traitement de cours qui comprend souvent un séjour à l'hôpital de 2 à 3 semaines et plusieurs mois de récupération. L'utilisation de melphalan oral en cures mensuelles est moins toxique mais est associée à un risque plus élevé de développer une leucémie.
De nouveaux médicaments actifs contre le myélome multiple (un autre trouble des plasmocytes anormaux), tels que le bortézomib ou le lénalidomide, sont également très efficaces contre la LA et se sont avérés bénéfiques chez les patients atteints d'une maladie récurrente. Souvent, ces médicaments sont inclus dans le prétraitement.
Actuellement, la majorité des patients n'utilisant pas de melphalan à haute dose avec support de cellules souches reçoivent une thérapie d'avant-garde. L'association du bortézomib, du cyclophosphamide et de la dexaméthasone est associée à une bonne tolérance et à des réponses rapides. Le traitement de l'amylose pour tout patient doit être personnalisé, en tenant compte des spécificités de la situation.
Les deux facteurs les plus importants dans la survie à long terme avec AL sont la présence/degré d'atteinte cardiaque et la réponse hématologique au traitement.
Il existe plusieurs nouveaux médicaments conçus pour stimuler la résorption de l'amyloïde des organes affectés. Leur utilisation peut permettre de traiter directement les organes malades. La plus avancée de ces études est celle de NEOD001, qui a montré un certain bénéfice chez les patients dont la maladie plasmocytaire sous-jacente était déjà traitée. Actuellement, la méthode est étudiée en association avec une thérapie à base de bortézomib au stade initial.
Thérapie d'entretienje (traitement de l'insuffisance cardiaque congestive, attention à la nutrition, traitement de la neuropathie autonome, etc.) est un élément très important de l'action médicinale. Compte tenu de la complexité de la maladie, il est recommandé que le traitement soit effectué dans un centre spécialisé dans l'amylose, ou au moins que le patient subisse une première évaluation dans un tel établissement médical avec poursuite du traitement sur le lieu de résidence.
Amylose familiale éliminé, si possible, en supprimant la cause première de la production anormale de TTP. Parce que le foie est la source dominante, la transplantation d'organes est actuellement le choix préféré chez des patients soigneusement sélectionnés dont la maladie est à des stades de développement tolérables. Tafamidis est un médicament récemment approuvé pour le traitement de la polyneuropathie amyloïde familiale. Ce médicament est testé dans des essais en cours pour d'autres formes de la maladie. Le patisiran et le revusiran sont également testés pour leur effet sur la forme ATTR de l'amylose, l'accent étant mis sur la réduction des niveaux de TTR qui forme l'amyloïde.
L'amylose est une maladie qui s'accompagne d'une violation du métabolisme des protéines et du fonctionnement du système immunitaire. La conséquence de tels troubles est l'amyloïde - dépôt intercellulaire de masses protéiques homogènes dans tous les organes du corps et ses tissus sans exception. En progressant, il déplace et remplace les cellules saines de l'organe atteint, entraînant ainsi la perte d'inactivité physique, ce qui provoque souvent le développement d'une dystrophie. Si l'amylose n'est pas traitée, elle contribue à la formation d'une défaillance multiviscérale, après quoi la mort peut survenir.
Les scientifiques médicaux ont découvert que l'amylose touche près de 1 % de la population mondiale. Dans le même temps, l'amylose secondaire survient plusieurs fois plus souvent que l'amylose primaire. Cette maladie peut être héréditaire. De tels cas ont été constatés parmi les représentants de nationalité arménienne et juive, ainsi que dans les pays méditerranéens.
Il existe 2 formes principales d'amylose :
- systémique (les tissus de la plupart des organes sont touchés);
- locale (lésions tissulaires d'un organe donné : reins, cœur, peau, etc.)
Un fait intéressant : les hommes souffrent d'amylose deux fois plus souvent que les femmes.
Classification de l'amylose
- Amylose primaire (idiopathique). Ses causes ne sont pas toujours connues. L'étude a révélé que les fibrilles amyloïdes sont une conséquence des chaînes d'immunoglobulines, appelées paramyloïdose. Les symptômes de l'amylose primaire peuvent inclure :
- la myasthénie grave, qui entraîne une atrophie musculaire ;
- douleur dans la cavité abdominale, accompagnée de diarrhée et de dyspepsie;
- violation du système génito-urinaire et ménopause;
- maladies des organes de la vision (rétinite, périartérite, etc.)
- Amylose secondaire (générale). Il survient dans l'environnement d'autres maladies, telles que la tuberculose, la bronchite, l'ostéomyélite, le paludisme, la syphilis, c'est-à-dire dans le contexte de maladies chroniques et d'infections. L'amylose générale s'accompagne d'une dégradation des tissus et d'une suppuration des organes. Les dommages aux intestins, au foie, à la rate et aux reins sont les plus courants. L'amylose secondaire passe longtemps inaperçue. Une légère faiblesse et une diminution de l'activité se produisent lorsque les reins sont touchés. Plus tard dans l'analyse, des protéines apparaissent dans l'urine. Au dernier stade, une insuffisance rénale se développe.
- amylose héréditaire. Elle est qualifiée par des mutations génétiques du système immunitaire, qui conduisent à la formation de cellules productrices d'amyloïde.L'amylose héréditaire peut inclure la fièvre, l'amylose neuropathique familiale, néphropathique et cardiopathique.
- Amylose cérébrale sénile. C'est le plus dangereux, car dans de telles circonstances, le muscle cardiaque, le pancréas, les poumons et le cerveau sont touchés. Elle est plus fréquente chez les personnes atteintes de la maladie d'Alzheimer.
- Amylose dans les tumeurs. La maladie se manifeste par le dépôt d'amyloïde dans les tissus de l'organe affecté, à la suite de quoi la croissance d'une tumeur maligne progresse. La cause de l'amylose dans les néoplasmes est le cancer de la glande endocrine et une tumeur de ses îlots.
Prévention de l'amylose
Cette terrible maladie peut être évitée en utilisant des régimes spéciaux et une variété de suppléments nutritionnels. Tout d'abord, il vous faut :
- réduire la consommation d'aliments riches en protéines. Cela s'applique principalement aux plats d'origine animale;
- augmenter la consommation de plats de poisson, d'aliments contenant des grains entiers, ainsi que de légumes, de fruits et de noix;
- renoncer aux produits laitiers, au sucre et aux aliments contenant de la caféine ;
- mangez plus d'aliments contenant de la vitamine C. Ce sont les agrumes, le chou, les tomates, les pommes, les groseilles, les pommes de terre au four et bien d'autres aliments.
Le traitement dépend du type de maladie. Si l'amylose est primaire, alors suivre un régime anti-protéiné conduira à un bon résultat. La chimiothérapie et l'utilisation de la colchicine peuvent également donner un résultat positif. L'amylose secondaire implique la nomination d'antibiotiques, de médicaments antiarythmiques et diurétiques qui agissent sur un organe spécifique.
Si les médicaments n'aident pas, une intervention chirurgicale est prescrite. De cette manière, une greffe de moelle osseuse, de rein ou de foie, une greffe de cœur et même l'implantation d'un stimulateur cardiaque peuvent être réalisées.
Dommages aux organes par l'amylose
- Dommages aux reins. Maladie fréquente et très dangereuse. Les symptômes de l'amylose des reins comportent 4 stades : latent, protéinurique, néphrotique et azotamique.
- Dommages au foie. La taille de l'organe augmente, ce qui peut être déterminé en sondant l'abdomen. Les lésions hépatiques amyloïdes se manifestent par une hypertrophie du foie, un ictère et une hypertension portale.
- amylose du coeur. Sa manifestation s'accompagne d'arythmie, d'essoufflement, d'évanouissement et de cardiomyopathie. Le dépôt amyloïde peut être présent à la fois dans le muscle cardiaque et dans ses membranes.
- amylose cutanée. Il contribue à une insuffisance de l'apport sanguin dans les vaisseaux, ce qui se traduit par une sécheresse et une desquamation de la peau. Une éruption cutanée peut apparaître sous la forme de petits boutons rouges.
- Maladie articulaire. Dans l'amylose des articulations, les mains et les pieds sont touchés en premier, puis les genoux et les coudes. Des mouvements douloureux, un œdème tissulaire, une température cutanée élevée au site de l'inflammation sont caractéristiques.
- Dommages musculaires. L'amylose de ces organes se manifeste par leur faiblesse, leur augmentation de taille et leur compaction. Accompagné de douleurs et de l'apparition de nodules denses. Si la maladie est déclenchée, les muscles sont comprimés, l'approvisionnement en sang est perturbé, ce qui entraîne leur mort.
- Maladie du système nerveux. L'amylose du système nerveux central est caractérisée par le dépôt d'amyloïde dans les tissus du cerveau, une conséquence directe de ce processus est une violation des capacités mentales du patient.
- Amylose du tractus gastro-intestinal. L'absorption des nutriments et des vitamines change. Cela se manifeste sous forme de diarrhée, de perte de poids soudaine, de fatigue, de troubles mentaux, ainsi que de perte de cheveux et d'anémie.
- Agrandissement de la rate. L'amylose de cet organe entraîne une augmentation de sa taille et le développement d'un hypersplénisme caractérisé par une anémie et une leucopénie.
- Amylose des poumons. Se produit avec une pneumonie, une pleurésie, un kyste. Le plus souvent situé sur les parois des vaisseaux des poumons et des bronches.
Le traitement de l'amylose ne peut être effectué qu'avec un examen précis du patient dans un hôpital.
Symptômes
Les signes d'amylose observés chez les personnes atteintes de cette maladie dépendent directement de l'organe atteint. À cet égard, il est impossible de distinguer un groupe spécifique de symptômes et, en fonction de l'amylose d'un organe particulier, des complexes de symptômes entiers sont distingués. Il convient également de noter que l'amylose généralisée primaire se caractérise par un polymorphisme du tableau clinique, car avec les intestins, le cœur, la peau, les reins, les systèmes endocrinien et nerveux peuvent être affectés.
Souvent, les premiers symptômes peuvent être une température corporelle élevée à long terme, une VS considérablement augmentée.
Lésions rénales dans l'amylose.
La localisation la plus fréquente est l'amylose des reins. Les symptômes de cette pathologie sont très divers et très dangereux sans la nomination d'un traitement adéquat. Le tableau clinique de l'amylose rénale se manifeste par certains symptômes d'insuffisance rénale chronique, à savoir :
- Modifications quantitatives des urines : oligurie (moins de 500 ml d'urine est excrétée par jour) et anurie (moins de 50 ml pendant la même période) sont observées.
- Troubles du bien-être général : apparition d'irritabilité, d'anxiété, de faiblesse et d'une fatigue accrue.
- L'apparition d'œdème - en raison d'une diminution de la concentration de protéines, il y a une transition de liquide dans les structures tissulaires, ce qui conduit à un œdème. Avec une progression prolongée de la maladie, du liquide s'accumule dans les cavités abdominale et thoracique, ainsi que dans la cavité péricardique, entraînant une ascite, un hydrothorax et un hydropéricarde.
- En raison de dommages au parenchyme du rein, une augmentation de la pression artérielle se produit, des arythmies et une hypertrophie du muscle cardiaque apparaissent.
- La violation de l'état mental et fonctionnel - qui se manifeste par l'effet néfaste de l'urée avec des reins non fonctionnels - entraîne une violation du fond mental, de l'insomnie, des troubles de la mémoire et des capacités mentales.
- Anémie : En raison de l'amylose des reins, la quantité requise d'érythropoïétine cesse d'être produite, ce qui stimule la production de globules rouges dans la moelle osseuse.
Dommages au foie dans l'amylose.
Avec les formes généralisées de la maladie, la soi-disant amylose hépatique se développe. Les symptômes sont dus au dépôt d'amyloïde dans le tissu hépatique et, par conséquent, à la compression des hépatocytes (cellules hépatiques) et des structures du lobule hépatique (veine, artère, voie biliaire). Avec l'accumulation d'amyloïde, la taille du foie augmente, l'hypertension portale se produit à la suite de la compression des vaisseaux sanguins. De plus, il y a une augmentation de la pression dans la veine porte, les veines de la rate, de l'œsophage et d'autres organes internes se dilatent. Tout cela se manifeste par l'apparition d'ascites, la survenue de saignements des veines dilatées de l'œsophage (souvent), des selles sanglantes (crayeuses), des vomissements avec du sang.
Lorsque les voies biliaires sont comprimées par l'amyloïde, un ictère survient (à la suite de l'accumulation de bilirubine), qui s'accompagne de fortes démangeaisons cutanées et d'un jaunissement de la sclérotique.
Dommages intestinaux dans l'amylose.
L'une des lésions est l'amylose intestinale. Les symptômes de cette forme sont caractérisés par:
- La diarrhée chronique - diarrhée constante, fréquente et prolongée - entraîne l'épuisement du corps et la personne commence à perdre du poids.
- Syndromes d'entéropathie exsudative et de malabsorption - le symptôme le plus courant de l'amylose intestinale - dans ce cas, une énorme quantité de protéines est perdue, qui migre du sang dans la lumière intestinale, se détachant. Le syndrome de malabsorption se caractérise par une évolution rapidement progressive. En relation avec l'exsudation (libération) simultanée de protéines dans la lumière intestinale, les troubles du métabolisme des protéines sous forme d'hypoprotéinémie persistante (diminution de la quantité de protéines dans le sang) et d'œdème prédominent. L'hypoprotéinémie dans ce cas est très difficile à corriger avec une thérapie de remplacement.
- Obstruction intestinale - se forme à la suite d'un dépôt local (local) d'amyloïde et du développement d'une formation semblable à une tumeur qui, en comprimant les anses intestinales, entraîne une violation de l'évacuation des matières fécales. Cette manifestation peut entraîner des complications graves, pouvant aller jusqu'à la septicémie.
- Les saignements gastro-intestinaux sont une manifestation assez courante de l'amylose intestinale, qui se caractérise par le développement de diverses formes d'anémie.
Dommages au système nerveux dans l'amylose.
Les symptômes initiaux les plus courants de l'amylose primaire du système nerveux comprennent une faiblesse générale, de la fatigue et une perte de poids assez rapide. Dans environ 40 % des cas, la polyneuropathie se développe avec une atteinte minimale des organes internes et, dans 15 % des cas, elle peut être la seule manifestation de la maladie. Mais dans 60% des cas, l'amylose systémique primaire conduit à une polyneuropathie dans le contexte de manifestations prononcées d'amylose d'une autre localisation (avec une amylose déjà existante du cœur, des reins, des intestins et autres). La polyneuropathie est de nature axonale, avec principalement des fibres minces myélinisées (myélinisées) et non myélinisées, et ressemble cliniquement à la polyneuropathie diabétique. Tout d'abord, les fibres sensorielles qui conduisent la sensibilité à la douleur et à la température, ainsi que les fibres autonomes, souffrent.
Les premiers symptômes de la polyneuropathie dans l'amylose du système nerveux sont :
- Engourdissement
- Dysesthésie (troubles sensoriels). La sensibilité dans ce cas souffre plus superficiellement que profondément
- Douleurs brûlantes dans les mains et les pieds.
La défaite des fibres et des ganglions autonomes par l'amylose se manifeste par une hypotension orthostatique, un pouls fixe, un dysfonctionnement érectile, une altération du trophisme cutané et une diminution de la transpiration.
À un stade ultérieur, une faiblesse musculaire et une amyotrophie (fonction musculaire et nutrition altérées) se développent. Dans le même temps, les troubles moteurs sont moins prononcés que les troubles sensibles.
Dommages aux tissus musculaires dans l'amylose.
La gravité des troubles du mouvement augmente si l'amylose affecte directement le tissu musculaire avec l'implication des muscles proximaux des membres.
La myopathie amyloïde se manifeste par des troubles plus qualitatifs principalement chez les enfants, associés à une faiblesse et à une fatigue de la couche musculaire déjà faiblement exprimée, résultant de leur compaction et de leur hypertrophie. Si l'amyloïde commence à se déposer dans les muscles de la langue, la soi-disant macroglossie se développe (une augmentation de la langue à une taille anormalement grande). Une dysphagie (incapacité à avaler) et un grossissement de la voix peuvent également apparaître. Chez un certain contingent de patients, une insuffisance respiratoire survient en raison de lésions des muscles respiratoires par l'amylose.
Lésions cutanées dans l'amylose.
Dans l'amylose primitive, on peut souvent noter l'une ou l'autre des éruptions cutanées : il s'agit le plus souvent de pétéchies, de purpura, de nodules et de plaques, alors qu'aucun critère général caractéristique de l'amylose cutanée ne peut être noté. Dans la forme de plaque nodulaire de l'amylose cutanée, on observe un grand nombre de plaques et de nœuds, situés principalement sur les tibias et les chevilles. Une caractéristique de cette forme d'amylose est la disposition symétrique de nodules isolés sur les deux jambes. Cette forme survient principalement chez les femmes.
Diagnostique
Méthodes modernes de diagnostic de l'amylose
L'amylose est une maladie qui se manifeste par une pathologie du métabolisme des protéines et des performances du système immunitaire. L'amyloïde (complexe protéine-saccharide), qui apparaît à la suite de ce trouble, peut se déposer dans les cellules de tous les tissus du corps. Avec le développement de la maladie, elle remplace progressivement les cellules saines et l'organe cesse de fonctionner. Dans une forme grave de pathologie, une défaillance multiviscérale survient (atteinte de 50 % ou plus des organes), entraînant la mort.
La forme héréditaire de la maladie est observée dans la région de la mer Méditerranée, ainsi que chez les personnes de nationalité juive et arménienne. Chez les hommes, cette maladie survient deux fois plus souvent.
Parmi les formes les plus courantes de la maladie, on peut nommer la néphropathie, dans laquelle des dépôts sont observés dans les reins, et des dépôts systémiques - amyloïdes se trouvent dans de nombreux organes.
L'amyloïde déposée dans différents organes a une structure différente. Il en existe environ 15 types au total, différant par leur structure et leur composition. Ils sont basés sur deux types :
- AA-amyloïde. Les maladies inflammatoires chroniques provoquent des taux plasmatiques élevés de protéine SAA et les maintiennent pendant une longue période. Avec un clivage protéique incomplet, une amyloïde AA fibrillaire se forme.
- AL-amyloïde. Des protéines de ce type apparaissent lors de la scission des amyloidoblastes (plasmocytes renaissants). Ce sont des composés immunoglobuliniques anormaux.
- L'apparition d'autres types d'amyloïde est déterminée par la forme de l'amylose.
Types de maladies
L'amylose survient comme une maladie indépendante et concomitante. Le diagnostic d'amylose distingue plusieurs types:
Primaire ou idiopathique. Avec ce type de dépôts se trouvent dans tous les organes internes. Il est impossible de déterminer la cause exacte de l'apparition. Il existe de multiples anomalies dans les cellules du système immunitaire avec l'accumulation d'amyloïde AL dans la peau, dans les tissus des systèmes musculaire, nerveux et cardiovasculaire. Sa cause peut être un plasmocytome (myélome multiple) - une pathologie tumorale maligne.
Symptômes de l'amylose primaire : - Myasthénie avec atrophie musculaire subséquente ;
- Dyspepsie et diarrhée;
- Pathologie du système génito-urinaire et reproducteur;
- Dommages aux organes de la vision.
Secondaire
C'est une complication de toute maladie inflammatoire. La raison de son apparition peut être:
- Maladies infectieuses chroniques : pyélonéphrite, tuberculose, bronchectasie, paludisme, syphilis ou lèpre (lèpre) ;
- Chronique maladies purulentes: ostéomyélite, ulcères purulents et plaies ;
- Colite ulcéreuse - inflammation du côlon;
- Lésions tumorales des organes hématopoïétiques : leucémie, lymphogranulomatose, etc. ;
- Pathologie rhumatologique: diverses arthrites et ainsi de suite.
L'amylose secondaire se forme dans les organes internes. Il y a une violation de l'activité des organes avec le plus grand dépôt d'amyloïde - dans la région des reins, de la rate, du foie ou des ganglions lymphatiques. À l'avenir, la lésion se propage à d'autres organes, suivie de la mort.
Héréditaire
Une forme similaire se forme en raison d'anomalies génétiques dans les cellules du système immunitaire, entraînant l'apparition d'amyloïdoblastes. Une pathologie similaire est diagnostiquée dans certains groupes nationaux ou dans une certaine zone géographique. La forme héréditaire comprend :
- Maladie périodique - fièvre familiale de la Méditerranée ;
- Amylose néphropathique ou anglaise familiale ;
- Amylose neuropathique héréditaire - portugaise, américaine ou finlandaise ;
- Cardiopathie héréditaire ou amylose danoise.
Sénile
Une approche systématique permet d'identifier cette pathologie chez les personnes de plus de 80 ans. Il comprend:
- Amylose cérébrale ou cérébrale. Diagnostiqué avec la maladie d'Alzheimer;
- amylose du coeur. Affecte le muscle cardiaque. Des dépôts se forment également dans les poumons, le foie et le pancréas.
tumeur
Dans ce cas, l'amylose se développe localement dans les organes présentant un processus malin prononcé. Elle est causée par un cancer médullaire de la thyroïde ou une tumeur des îlots pancréatiques.
Hémodialyse
Avec l'hémodialyse, prescrite aux patients souffrant d'insuffisance rénale, la teneur en B 2 -microglobuline dans le sang augmente progressivement. Cette protéine, lorsqu'elle interagit avec les nucléoprotéines, se dépose dans les tissus des reins.
Diagnostic de l'amylose
Pour diagnostiquer l'amylose, un patient se voit attribuer un certain nombre d'études différentes. Il s'agit d'un test sanguin et urinaire général, d'une biochimie sanguine, d'une échographie des organes internes, d'une biopsie et d'un test génétique.
Analyse sanguine générale
Identifie les anomalies spécifiques à l'amylose. Dans les derniers stades de la maladie, cette étude permet d'identifier l'organe lésé.
Analyse d'urine générale
Le diagnostic d'amylose des reins montre la probabilité de développer des processus inflammatoires se produisant dans les reins.
Avec la pathologie rénale, les éléments suivants sont détectés:
- Protéinurie - la teneur en protéines dans l'urine supérieure à 3 g / l;
- Hématurie - détection de globules rouges dans l'urine;
- Leucocyturie - la présence de leucocytes dans l'urine;
- Cylindrurie - le contenu dans l'urine des cylindres formés au cours de l'amylose à partir de protéines, de cellules épithéliales des reins, de leucocytes et d'érythrocytes;
- Diminution de la densité de l'urine.
Chimie sanguine
Il permet d'évaluer l'état général du corps et d'établir la cause de l'amylose. Cette analyse détermine :
- Protéines de phase générale produites par le foie ou certains globules blancs lors d'un processus inflammatoire. Une attention particulière doit être portée à la quantité de fibrinogène.
- Les tests hépatiques indiquent l'état de cet organe.
- Une augmentation du taux de cholestérol est un signe de syndrome néphrotique.
- Une diminution des taux de protéines indique un syndrome néphrotique ou une insuffisance hépatique.
- Une augmentation de la quantité d'urée et de créatinine est un indicateur de dysfonctionnement rénal dans l'amylose.
Échographie
Cette méthode permet de déterminer la structure et la structure des tissus des organes internes, le degré et la propagation des processus pathologiques.
Le diagnostic de l'amylose par échographie montre:
- Compactage et modification de la taille des reins;
- La présence de kystes dans les reins;
- Compactage et élargissement de la rate et du foie, accompagnés d'une pathologie du flux sanguin;
- Hypertrophie du muscle cardiaque;
- La présence de dépôts amyloïdes dans les parois des principaux vaisseaux sanguins ;
- Une augmentation du volume de liquide dans diverses cavités du corps - ascite, hydropéricarde ou hydrothorax.
Biopsie
Retrait d'un petit morceau de tissu pour examen à l'aide de méthodes spéciales. Son utilisation permet de diagnostiquer l'amylose dans 90% des cas. Le tissu du muscle, des organes internes, ainsi que la membrane muqueuse sont prélevés pour la recherche.
recherche génétique
Elle est réalisée avec la probabilité de développer une amylose héréditaire. Le matériel génétique du patient est prélevé pour l'étude, qui est vérifiée pour la présence d'anomalies génétiques dans certains chromosomes. Si une pathologie est détectée, il est recommandé que tous les parents de sang du patient subissent un examen pour identifier cette maladie en eux.
Traitement
Le traitement de l'amylose est symptomatique et de soutien - il est impossible de guérir complètement la maladie. L'hospitalisation est recommandée pour le patient, car avec des signes prononcés de la maladie, une forte détérioration dangereuse de l'état et l'apparition de complications sont possibles. Après stabilisation du patient, le traitement est effectué à domicile.
Si la maladie a une forme secondaire et est à un stade précoce, le traitement de la maladie sous-jacente est efficace. Dans ce cas, les symptômes douloureux de l'amylose s'affaiblissent.
Le traitement médical de l'amylose vise à ralentir ou à limiter au maximum la formation d'amyloïdes et à atténuer l'évolution de la maladie. Pour cela, des médicaments sont utilisés:
- anti-inflammatoire stéroïdien;
- antitumoral et aminoquinoline, qui inhibent la synthèse des protéines;
- antigoutte (avec une forme héréditaire de la maladie), qui ralentit la vitesse de formation des leucocytes.
Si l'amylose est diagnostiquée, le régime est prescrit dès qu'ils en ont connaissance: il aide à protéger le cœur, les reins et les autres organes affectés des produits métaboliques et normalise l'équilibre eau-sel, égalise la tension artérielle. La nourriture doit être prise souvent, toutes les trois heures, en petites portions. L'accent doit être mis sur les bouillons de légumes, les fruits et légumes frais, les viandes maigres, le poisson et les produits laitiers. Il est recommandé d'éviter les bouillons forts à base de poisson et de viande, l'utilisation de fromage, de jaune d'œuf, de pâtisseries, de café et d'alcool n'est pas souhaitable. Il est obligatoire de limiter la consommation de sel (jusqu'à 2 g par jour) pour prévenir les œdèmes.
Certains patients pensent que le traitement à jeun peut être une panacée pour l'amylose, mais les médecins dissipent ces idées : le patient a besoin d'un repas régulier et son manque menace de complications.
Dans le même temps, ils ne s'opposent pas à ce que le patient utilise des remèdes populaires sous la forme de préparations à base de plantes ayant des effets anti-inflammatoires et diurétiques, mais ils avertissent qu'ils ne peuvent servir que de mesures auxiliaires, mais ne remplacent pas le traitement principal.
La maladie progresse et, souvent, la prise de médicaments seuls ne suffit pas.
Types de dialyse pratiquées pour l'amylose
Si un patient est diagnostiqué avec une amylose des reins, le traitement, afin d'éviter le développement d'une insuffisance rénale, est mis en œuvre par dialyse. Il assure l'élimination des produits toxiques de l'organisme et contribue à la normalisation de l'équilibre eau-sel, acido-basique. Dans l'amylose, deux types de dialyse peuvent être pratiqués :
- hémodialyse;
- péritonéale.
Hémodialyse
La procédure est réalisée à l'aide d'une membrane artificielle semi-perméable qui sépare le sang du patient du dialysat. Le sang du patient contient une forte concentration de substances nocives (urée, protéines, toxines), qui ne sont pas en solution. À la suite de la diffusion, ces substances pénètrent dans la membrane, saturant le liquide avec une concentration plus faible. Les éléments vitaux nécessaires (ions sodium, magnésium, calcium, potassium, chlore) sont reconstitués à l'aide de dialysat à la concentration requise afin qu'ils ne restent pas dans le sang. Ainsi, le dispositif remplace un rein non fonctionnel. Si une personne souffre d'un œdème des articulations, des poumons (un œdème similaire peut également se trouver sous la peau ou autour du cœur), alors en raison du passage de l'excès de liquide dans la solution en raison de la différence de pression, ils passent.
Dialyse péritonéale
Le traitement de l'amylose rénale est efficace avec la dialyse péritonéale : les médecins pensent que l'élimination de la microglobuline B2, qui affecte la formation d'amyloïdes, est plus efficace qu'avec l'hémodialyse. Le principe de cette procédure est similaire au principe de l'hémodialyse, mais le péritoine agit comme une membrane à travers laquelle s'effectue l'excrétion des produits métaboliques. C'est le nom de la fine membrane qui recouvre la surface interne de la cavité abdominale et de ses organes.
Cette procédure nécessite la mise en place d'un cathéter dans la cavité pelvienne. Cette manipulation est réalisée à l'aide d'une intervention chirurgicale. La partie externe du tube est sortie sous la peau devant ou sur le côté de la cavité abdominale.
Grâce à cela, le traitement de l'amylose par dialyse devient possible à domicile : une procédure qui doit être effectuée tous les jours peut être prise en charge par le patient lui-même. Ceci est considéré comme un gros plus par rapport à l'hémodialyse. Le cathéter est bien fixé, le risque d'infection dans ce cas est minime.
Le dialysat est introduit dans la cavité abdominale à l'aide d'un cathéter et les toxines commencent à y pénétrer à partir des vaisseaux de la paroi péritonéale (cette zone est bien irriguée). La solution est contaminée après quelques heures, elle est donc remplacée par la dose suivante (généralement 2 litres). Pendant que le liquide est à l'intérieur, aucune autre mesure n'est requise - le patient peut vaquer à ses activités quotidiennes.
Greffe de tissus ou d'organes
En cas d'apparition d'une défaillance d'organe, la transplantation d'un organe malade (s'il s'agit d'un rein ou d'un cœur) ou d'un tissu (lorsque le foie, la peau est touché) est nécessaire. Si une amylose se développe dans la rate, elle doit être retirée.
Après une greffe de donneur, le patient prend des médicaments immunosuppresseurs pour le reste de sa vie - cela est nécessaire pour que son propre corps ne rejette pas le tissu ou l'organe greffé. Cependant, l'utilisation d'un organe donneur n'est pas une panacée, car dans la plupart des cas, une rechute de la maladie est possible, ce qui aggrave le pronostic.
La transplantation d'organes est réalisée à la fois dans des cliniques nationales et à l'étranger (en Allemagne, en Israël), où ce domaine de la médecine est particulièrement développé.
Médicaments
Au début, lorsque la maladie ne s'est manifestée que, les médicaments à base d'aminoquinoléine aident. Seul un médecin peut prescrire ces médicaments. Rappelez-vous également qu'ils ne sont pas efficaces dans le cours aigu ou dans les derniers stades de la maladie.
Si la pression artérielle a augmenté de manière significative à cause de l'amylose, des médicaments sont prescrits pour la normaliser. Avec la formation d'œdème, des diurétiques d'un certain dosage sont utilisés, en fonction de la gravité du syndrome néphrotique. Si le patient a une diminution de l'hémoglobine, vous devez prendre des médicaments contenant du fer afin de normaliser la composition du sang.
Dans le traitement de cette maladie, des immunosuppresseurs sont souvent prescrits. Dans l'amylose primaire, il est recommandé de prendre Melphalan, Prednisolone. En présence d'amylose AA, la Colchicine, l'Unithiol sont prescrits. Pour le traitement de nombreux patients, des médicaments désensibilisants sont utilisés - Suprastin,
Pipolfen. Dans l'amylose secondaire, qui apparaît comme une complication de la polyarthrite rhumatoïde, des anti-inflammatoires spécifiques sont prescrits.
Prendre des médicaments aux premiers stades de la maladie
Dans le traitement de l'amylose, on utilise des médicaments de la série des aminoquinoléines, qui visent à prévenir la formation d'amyloïde dans les tissus corporels. Ils doivent être pris pendant 2-3 mois à plusieurs années pour prévenir les effets secondaires.
Les médicaments à base d'aminoquinoline peuvent nuire à la vision. Par conséquent, dans le processus de traitement, il est nécessaire de subir périodiquement un examen par un ophtalmologiste afin d'identifier les violations à temps. D'autres effets secondaires sont également possibles - diarrhée, vomissements, psychose, éruptions cutanées. Avec un effet négatif prononcé des médicaments de ce groupe, ils sont annulés.
Colchicine pour l'amylose
En utilisant la colchicine pendant longtemps, vous pouvez oublier les nouvelles attaques de la maladie, c'est un excellent prophylactique. Si ce médicament est pris à vie, vous pouvez obtenir les résultats suivants :
- longue rémission;
- la transplantation rénale prévient la récurrence de la maladie;
- disparition complète du syndrome néphrotique;
- élimination de la protéinurie si les reins du patient fonctionnent bien.
La dose quotidienne recommandée du médicament pour l'amylose est de 1,8 à 2 mg. En présence d'insuffisance rénale, la quantité de colchicine est légèrement réduite. Ce médicament est suffisamment sûr pour les humains. Il peut être pris pendant une longue période. Dans de rares cas, des effets secondaires sont possibles sous forme d'indigestion, de douleurs abdominales. En présence de ces symptômes, il n'est pas nécessaire d'annuler le traitement par Colchicine. Dans quelques jours, tout devrait revenir à la normale. Pour soulager l'état du patient, une prise supplémentaire de préparations enzymatiques est autorisée pendant 2-3 jours.
Médicaments pour le traitement de l'amylose causée par la polyarthrite rhumatoïde
Dans l'amylose secondaire, le Dimexide est utilisé. Cet agent anti-inflammatoire doit être pris par voie orale en une quantité de 1 à 5 ml. En présence d'amylose des reins, le médicament est prescrit avec beaucoup de soin, en commençant par la dose minimale. L'ensemble du processus de traitement doit être sous la supervision d'un médecin.
Traitement de l'amylose primaire
Pour aider un patient atteint d'amylose primaire, on utilise Melphalan, qui est associé à un autre médicament, la prednisolone. La durée du traitement est longue: pas moins d'un an. Les médicaments sont pris pendant 4 à 7 jours, après quoi vous devez faire une pause de 28 à 45 jours. Le nombre de cures est déterminé par le médecin.
La posologie des médicaments dépend du poids de la personne. Pour 1 kg de poids, 0,2 mg de Melphalan et 0,8 mg de Prednisolone sont prescrits. Cette posologie est indiquée pour la prise de médicaments pendant 24 heures.
L'effet positif du traitement avec ces médicaments:
- une diminution significative des protéines dans l'urine;
- normalisation de la teneur en créatinine dans le sang du patient ;
- amélioration de la circulation sanguine;
- diminution de la quantité d'immunoglobuline.
Avec l'amylose primaire, il n'est souvent pas possible de mener un traitement à long terme avec ces médicaments, car l'état d'une personne peut se détériorer rapidement. Ce schéma est acceptable pour les patients sans insuffisance rénale sévère.
Remèdes populaires
Le traitement de l'amylose avec des remèdes populaires peut avoir un effet positif sur l'état de santé, mais ne doit être effectué qu'avec le consentement du médecin traitant.
Dans de nombreuses maladies, il est obligatoire de suivre un régime alimentaire, et l'amylose ne fait pas exception à cette règle. Il est nécessaire d'exclure les protéines et le sel de votre alimentation, car. ils affectent l'insuffisance rénale et cardiaque. Il est souhaitable d'augmenter la consommation d'aliments contenant de l'amidon, de l'acide ascorbique, des sels de potassium. Ces aliments comprennent les produits de boulangerie, les pommes de terre, les céréales, les poivrons, les légumes verts (épinards, brocoli, choux de Bruxelles, aneth), les agrumes, l'ail, les carottes, le foie, le poisson et les produits laitiers.
La médecine traditionnelle recommande de manger du foie cru dans le traitement de l'amylose, qui contient une grande quantité de substances utiles et nécessaires à notre organisme (acide ascorbique, tocophérol, vitamines B, carotène, axérophtol). Le foie contient également une riche composition d'éléments minéraux: P, Fe, Zn, Cu. À la suite de l'apport quotidien de foie cru (pendant un an et demi à deux ans), on peut observer une amélioration du fonctionnement des reins, du tractus gastro-intestinal et du système nerveux.
Pour renforcer le système immunitaire, on utilise des plantes médicinales qui ont des propriétés anti-inflammatoires. Les remèdes suivants sont populaires dans la médecine traditionnelle :
- Infusion thérapeutique de camomille, bourgeons de bouleau, immortelle, millepertuis. Ces plantes sont mélangées et le mélange est placé dans un thermos et versé avec de l'eau bouillante, après quoi il est autorisé à infuser, filtré et consommé la nuit.
- Décoction d'ortie (destiné à nettoyer le sang, a également un effet anti-inflammatoire).
- Le thé est infusé à partir de fleurs et de feuilles d'ortie, qui sont bouillies avant d'être bues.
- Thé de fraises, menthe, millepertuis. Utilisé comme tonique général.
Teinture alcoolique d'herbe d'avoine. On pense que ce médicament a un effet bénéfique sur l'activité fonctionnelle du cœur, des vaisseaux sanguins et du système nerveux. Lors de la cuisson, l'herbe est broyée, versée dans une bouteille et versée avec de l'alcool, après quoi elle est placée dans un endroit sombre (il est recommandé de la filtrer périodiquement). Avant utilisation, diluer la teinture avec de l'eau.
Les informations sont fournies à titre indicatif uniquement et ne constituent pas un guide d'action. Ne vous soignez pas vous-même. Aux premiers symptômes de la maladie, consultez un médecin.
Une maladie causée par une violation du métabolisme des protéines, dans laquelle la formation et le dépôt dans divers tissus et organes d'une substance protéique-polysaccharidique spécifique - l'amyloïde.
Développement de la maladie
L'amylose se développe (ce que c'est - nous l'avons déjà découvert) en violation de la synthèse des protéines dans le système réticulo-endothélial. Il y a une accumulation de protéines anormales. Ces protéines sont essentiellement des auto-antigènes et provoquent, par analogie avec les allergies, la formation d'auto-anticorps.
Ensuite, ces anticorps réagissent avec les antigènes et les protéines grossièrement dispersées précipitent. C'est ainsi que se forme l'amyloïde. Cette substance se dépose sur les parois vasculaires et divers organes. S'accumulant progressivement, l'amyloïde entraîne la mort de l'organe.
Types d'amylose. causes
Il existe plusieurs types d'amylose. Les causes du développement de la maladie dépendent directement du type d'amylose. Ce que c'est? La classification est effectuée en fonction de la protéine principale qui compose les fibrilles amyloïdes. Voici les types de cette maladie.
- Amylose primaire (amylose AL). Avec son développement, des chaînes légères anormales d'immunoglobulines apparaissent dans le plasma sanguin, capables de se déposer dans divers tissus du corps. De la même manière, les plasmocytes changent avec la macroglobulinémie de Waldenström, l'hypergammaglobulinémie monoclonale.
- Amylose secondaire (amylose AA). Dans ce cas, il y a un excès de sécrétion de protéine alpha-globuline par le foie. C'est une protéine de phase aiguë qui est synthétisée au cours d'un processus inflammatoire chronique. Cela est possible avec diverses maladies, par exemple la polyarthrite rhumatoïde, le paludisme, l'ostéomyélite, la lèpre, la tuberculose.
- Amylose familiale (amylose FA). Il s'agit d'une forme héréditaire de la maladie avec un mécanisme de transmission autosomique récessif. On l'appelle aussi fièvre intermittente méditerranéenne ou polysérite paroxystique familiale. Cette maladie se traduit par des accès de fièvre, la survenue de douleurs abdominales, des éruptions cutanées, de l'arthrite et de la pleurésie.
- Amylose de dialyse (AH-amylose). Il est associé au fait que la protéine bêta-2-microglobuline MHC chez les personnes en bonne santé est utilisée par les reins, et pendant l'hémodialyse, elle n'est pas filtrée et s'accumule donc dans le corps.
- Amylose AE. Il se développe dans certaines formes de cancer, comme le cancer de la thyroïde.
- Amylose sénile.

Symptômes
Lors d'un diagnostic d'amylose, les symptômes dépendent de l'emplacement des dépôts. Si le tractus gastro-intestinal est affecté, il peut y avoir une hypertrophie de la langue, des troubles de la déglutition, de la constipation ou de la diarrhée. Des dépôts de type tumeur amyloïde dans l'intestin ou l'estomac sont parfois possibles.
L'amylose intestinale s'accompagne d'une sensation de lourdeur et d'inconfort, il peut y avoir des douleurs modérées dans l'abdomen. Si le pancréas est affecté, les mêmes symptômes sont présents que pour la pancréatite. Lorsque le foie est endommagé, son augmentation est observée, des nausées, des éructations, des vomissements, une jaunisse apparaissent.
L'amylose du système respiratoire se manifeste comme suit:
- voix rauque;
- symptômes de bronchite;
- amylose pulmonaire de type tumoral.
Avec l'amylose du système nerveux, les symptômes suivants peuvent être observés:
- sensations de picotements ou de brûlures dans les extrémités, engourdissements (polyneuropathie périphérique);
- maux de tête, vertiges;
- troubles sphinctériens (incontinence urinaire, selles).
Amylose - ce que c'est, nous avons examiné les causes de son apparition et ses symptômes. Voyons maintenant comment cette maladie est diagnostiquée et quelles méthodes de traitement existent.

Diagnostique
Avec une maladie comme l'amylose, le diagnostic est complexe. Des études de laboratoire et de matériel sont confiées.
Dans les études de laboratoire sur le test sanguin général, une augmentation de la VS, des leucocytes et une diminution des plaquettes sont observées. Dans l'analyse générale de l'urine, il y a des protéines, dans le sédiment, il y a des cylindres, des leucocytes et des érythrocytes. Le coprogramme contient une grande quantité d'amidon, de graisse et de fibres musculaires. Dans la biochimie du sang présentant des lésions hépatiques, on trouve une teneur accrue en cholestérol, en bilirubine et en phosphatase alcaline.
Dans l'amylose primaire, une teneur élevée en amyloïde se trouve dans l'urine et le plasma sanguin. Dans le secondaire lors des tests de laboratoire, on trouve des signes d'un processus inflammatoire chronique.
D'autres mesures de diagnostic sont également effectuées:
- examen radiologique;
- échocardiographie (en cas de suspicion de maladie cardiaque);
- tests fonctionnels avec des colorants;
- biopsie d'organe.

Traitement
Cette maladie est traitée en ambulatoire. L'amylose, dans laquelle des conditions graves sont observées, par exemple dans l'insuffisance rénale chronique ou l'insuffisance cardiaque grave, est traitée dans un hôpital.
Dans l'amylose primaire, au stade initial, des médicaments tels que la chloroquine, le melphalan, la prednisolone, la colchicine sont prescrits.
Dans l'amylose secondaire, la maladie sous-jacente est traitée, par exemple l'ostéomyélite, la tuberculose, l'empyème pleural, etc. Souvent, après son traitement, tous les symptômes de l'amylose disparaissent.
Si la maladie se développe à la suite d'une hémodialyse rénale, un tel patient est transféré en dialyse péritonéale.
En cas de diarrhée, des astringents sont utilisés, par exemple, "Bismuth Subnitrate" ou des adsorbants.
Un traitement symptomatique est également utilisé :
- médicaments qui réduisent la tension artérielle;
- vitamines, diurétiques;
- transfusion de plasma, etc.
De plus, un traitement chirurgical peut également être utilisé. L'amylose de la rate peut régresser après le retrait de l'organe. Dans la plupart des cas, cela conduit à une amélioration de l'état des patients et à une diminution de la formation d'amyloïde.
Nutrition
L'amylose nécessite une alimentation constante. Avec le développement de l'insuffisance rénale chronique, la consommation de produits salés et protéiques tels que la viande, le poisson et les œufs doit être limitée. Si une maladie chronique se développe, les aliments salés, fumés et marinés doivent être exclus de l'alimentation.

Cette maladie est aussi appelée cardiopathie amyloïde. Avec son développement, un dépôt amyloïde peut se produire dans le myocarde, le péricarde, l'endocarde ou sur les parois de l'aorte et des vaisseaux coronaires. La cause de tels dommages au cœur peut être une amylose primaire, secondaire ou familiale. Souvent, l'amylose du cœur n'est pas une maladie isolée et elle se développe en parallèle avec l'amylose des poumons, des reins, des intestins ou de la rate.
Symptômes de l'amylose du coeur
Souvent, les symptômes de cette maladie sont similaires à ceux de la cardiopathie hypertrophique ou de la maladie coronarienne. Au stade initial, les symptômes ne sont pas clairement exprimés. Il peut y avoir de l'irritabilité et de la fatigue, une perte de poids, un gonflement des tissus et des étourdissements.

Une forte détérioration survient généralement après toute situation stressante ou une infection respiratoire. Après cela, des douleurs cardiaques apparaissent généralement en fonction du type d'angine de poitrine, d'arythmies, d'œdème prononcé, d'essoufflement, d'hypertrophie du foie. La tension artérielle est généralement basse.
La maladie progresse rapidement et sa caractéristique est la résistance (résistance) au traitement en cours. Dans les cas graves, les patients peuvent présenter une ascite (accumulation de liquide ou épanchement péricardique). En raison des infiltrats amyloïdes, une faiblesse du nœud sinusal et une bradycardie se développent. Cela peut entraîner une mort subite.

Prévision
Avec l'amylose du cœur, le pronostic est défavorable. L'insuffisance cardiaque dans cette maladie progresse régulièrement et la mort est inévitable. Il n'y a pas de centres spécialisés traitant de ce problème en Russie.